Review - (2021) Volume 5, Issue 4
Received: 29-Jun-2021
Published:
21-Jul-2021
, DOI: 10.37421/2684-4559.2021.5.154
Citation: Musher Daniel M, et al. "Contribution of Animal Studies to the Understanding of Infectious Diseases". Clin Infect Dis 5 (2021) doi: 154
Copyright: © 2021 Musher DM, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Experiments in animals have played an integral role in furthering basic understanding of the pathophysiology, host immune response, diagnosis, and treatment of infectious diseases. However, competing demands of modern-day clinical training and increasingly stringent requirements to perform animal research have reduced the exposure of infectious disease physicians to animal studies. For practitioners of infectious diseases and, especially, for contemporary trainees in infectious diseases, it is important to appreciate this historical body of work and its impact on current clinical practice. In this article, we provide an overview of some major contributions of animal studies to the field of infectious diseases. Areas covered include transmission of infection, elucidation of innate and adaptive host immune responses, testing of antimicrobials, pathogenesis and treatment of endocarditis, osteomyelitis, intra-abdominal and urinary tract infection, treatment of infection associated with a foreign body or in the presence of neutropenia, and toxin-mediated disease.
Experimental animal model • Foreign body • Endocarditis • Osteomyelitis • Urinary tract infection
The modern training of infectious disease physicians offers little opportunity to work with experimental animal models. Competing demands of clinical training requirements, acquisition of other research methodologies, more stringent requirements of institutional review boards and distancing of animal research facilities from clinical settings, have made it difficult to begin even a pilot study involving animal experimentation. An emerging generation of infectious disease specialists may have little appreciation for how experimental work in animals has helped to shape the field of infectious diseases. The present submission attempts to give a brief overview of ways in which experiments in animal models have greatly broadened the understanding of our chosen field.
Tuberculosis and Koch’s Postulates
Any discussion of the importance of animal models in human infectious diseases must begin with the work of Robert Koch. True, Pasteur had recognized, in 1862 that, after burial of sheep that died of anthrax, microorganisms survived in soil, were carried to the surface by earthworms where they spread the disease to healthy animals. Within three years, Villemin [1] reported that sputum from patients with tuberculosis could transmit infection to rabbits, work that went largely unrecognized by the German scientific establishment [2], perhaps because of nationalist rivalries. But, in 1882, in a lecture that was literally stunning (there was no applause afterwards), Koch [3] showed that:
(a) With appropriate staining, microorganisms could be seen microscopically in all tuberculous lesions;
(b) These organisms could be cultivated in vitro on appropriate medium;
(c) Injection of cultivated organisms into guinea pigs caused tuberculosis with typical tuberculous lesions; and
(d) The causative organism could then be isolated from these lesions. These so-called Koch’s postulates, based on experiments in rodents, established the basis for the modern understanding of infectious diseases.
Only in the middle of the twentieth century did Riley et al., [4] use animal experiments to establish the principle of airborne transmission of infection, as opposed to droplet transmission or direct contact. This work was done by piping air from hospital rooms that housed tuberculous patients across the cages of guinea pigs. Interestingly after a short course of therapy, even though organisms were still present in patients’ sputum, there was no longer a risk of contagion, a principle that may yet to be fully appreciated by experts in infection control.
Pneumococcus and Humoral Immunity
In 1880, the organism we have come to call Streptococcus Pneumoniae was discovered in animal studies by two investigators in different countries, in nearly the identical fashion and entirely by chance. Sternberg, who was studying malaria in New Orleans, injected his own saliva into rabbits, putatively as a negative control, and Pasteur who was studying rabies in Paris injected saliva from a child who had rabies into a rabbit in an attempt to transfer rabies. The rabbits promptly sickened and died, and the investigators isolated organisms from the bloodstream of infected animals. Pasteur cultivated these organisms in broth, identified them as cocci and showed that Injection into other rabbits was lethal.
In the 1890s, Felix and Georg Klemperer showed that repeated injection of killed pneumococci immunized rabbits against pneumococcal challenge. When they transferred serum from immune into non-immune rabbits, the recipients were no longer susceptible to challenge with the immunizing strain [5]. These investigators then found another pneumococcal isolate against which these rabbits were not protected. Repeating the same steps as initially, they rendered rabbits immune to this new isolate but not to the original one. Thus, in a series of experiments in rabbits, the Klemperers (1) identified two different types of S. pneumoniae, (2) showed that serum contained a transferable protective substance, thereby establishing the concept of humoral immunity, and (3) paved the way for studies of pneumococcal vaccination, begun in humans in 1910 by Wright [6].
In the 1920’s, Felton showed that the soluble carbohydrate of pneumococci was immunogenic and specifically protected mice against pneumococcal challenge [7]. These findings led directly to the isolation and purification of pneumococcal capsular polysaccharide for use as an effective vaccine in humans [8].
BCG, Brucella and Cellular Immunity
In 1919, Calmette and Guerin found that vaccinating guinea pigs with a live, attenuated strain of M. Tuberculosis (BCG), protected them against challenge with unattenuated tubercle bacilli. Less than a year later, they reported efficacy of BCG in protecting humans against disseminated infection. This immunity was not transferred by serum. Mackaness [9] reported that activation of macrophages was responsible, and that this activation was not specific to the challenging organism [10]; mice infected with BCG were protected against challenge with unrelated organisms such as Brucella and Salmonella as well as M. Tuberculosis. Important later experiments in rodents showed that humoral immune responses were governed by B-lymphocytes and cell-mediated responses by T-lymphocytes [11, 12].
DNA as the Genetic Principle
Griffith [13] injected pneumococci that lacked capsules subcutaneously into mice together with killed encapsulated pneumococci. The mice rapidly sickened and died, and encapsulated pneumococci were recovered from their blood stream. Griffith called this phenomenon reversion, attributing it to bacterial proteins. More than a decade later, in similar experiments in mice, Avery, MacLeod and McCarty [14] used purified DNA to transform unencapsulated pneumococci to virulent, encapsulated forms, thereby establishing the role of DNA as the genetic principle.
Testing of Antimicrobials
In the early 1930s, Domagk documented the beneficial effect and potential toxicity of sulfanilamide in streptococcal infections in mice. Whitby extended this work to S. Pneumoniae using a modified form of the drug [15]. These experiments established a pattern for development of antimicrobial drugs, in those days leading much more directly to their use in humans than at present.
More recently, a model of soft tissue infection in neutropenic mice elucidated important principles of infectious diseases by documenting the:
(1) Relevance of bactericidal vs bacteriostatic antibiotic activity in neutropenia [16];
(2) Relation of pharmacokinetics to outcomes of therapy [16];
(3) So-called ‘post-antibiotic effect’ of aminoglycosides in vivo [17]; and
(4) Superiority of continuous antibiotic infusion for treating neutropenic subjects [18].
Infection and Foreign Bodies
The difficulty of curing an infection in the presence of a foreign body was known from the beginning of the antibiotic era. The principle underlying management was simply to remove the foreign body. But as implants became more common, it became clear that this often could not be done. When infections developed at the site of an implant, infectious disease specialists recommended months, years or even lifetime treatment with the same cell-wall active antibiotics used to treat acute infections. At first, persistence of bacteria in the presence of bactericidal concentrations of antibiotics was attributed to survival within polymorphonuclear leukocytes [19]. The success of rifampin in killing S. aureus under such circumstances was attributed to the penetration of this drug into PMN [20], although emergence of resistant variant strains was also recognized as a contributor to bacterial survival [21]. Demonstration of biofilm, complex bacterial structures on the surface of implanted objects [22, 23], suggested that the failure to eradicate bacteria was due to poor penetration. However, Darouiche et al., [24] showed that antibiotics penetrated biofilm and suggested that altered bacterial metabolism was responsible for the failure to be eradicated by antibiotics; Costerton et al., [25] proposed that these metabolic changes mandated different antibiotic approaches.
Accordingly, experiments were begun in animals to study infections associated with implanted devices. Zimmerli et al., [25] developed a model in which cages containing coverslips were implanted subcutaneously into guinea pigs and later inoculated directly with bacteria. Chuard et al., [26] used this model to show that the minimum bactericidal concentration of antibiotics for bacteria was greatly increased in the biofilm state and that, whereas vancomycin or fleroxacin (a fluoroquinolone) failed to clear bacteria, these two drugs in combination with rifampin sterilized > 90% of coverslips [27]. Importantly, the investigators emphasized that these results could not have been predicted from in vitro studies. As a result of these and related experiments in animals, combinations of non-beta lactam antibiotics have become central to treating infections associated with implants and, by extension, osteomyelitis (see below). Zimmerli et al., [28] used this animal model to demonstrate what has become another important clinical principle, namely, that implanted foreign bodies can become infected during bacteremia.
Osteomyelitis
To some extent, the challenges of treating chronic osteomyelitis can be attributed to the same phenomenon, because devitalized bone acts as a foreign body surrounded by infection. In the late 1800s, Rodet [29] and Lexer [30] injected Staphylococci intravenously in rabbits resulting in discrete bone abscesses, but this model did not mimic the diffuse long bone disease seen in humans [31]. In 1926, Wilensky proposed vascular thrombosis as a key pathogenic factor in the development of osteomyelitis [32]. It took 15 years before Scheman and colleagues injected sodium morrhuate (a sclerosing agent designed to cause microvascular occlusion) into the tibial metaphysis of rabbits followed by injection of staphylococci either directly into bone or intravenously [33], resulting in progressive radiographic changes of osteomyelitis and histologic findings of suppuration and sequestration. Interestingly, little evidence of microvascular occlusion was seen leading Scheman to propose that direct trauma to the bone matrix was the mechanism of morrhuate. As later shown in a variety of animal models, this disruption of bone environment could be achieved by numerous methods including injection of other sclerosing agents (e.g. arachidonic acid) [34], open fracture [35], or placement of foreign bodies (wires [36], screws, intramedullary nail [37] or joint implants [38]). Importantly, such manipulations of the bone environment also permitted establishment of infection at several log-fold lower inocula of bacteria.
In 1970, Norden and Kennedy modified Scheman’s model, reaffirming the necessity of morrhuate for development of osteomyelitis, and observing infected rabbits for up to 6 months after infection [39]. The uniformity, reliability and longevity of this osteomyelitis model paved the way for experiments evaluating antimicrobial therapy particularly focusing on Staphylococcus. Aureus and Pseudomonas. Aeruginosa osteomyelitis [40],. These experiments underscored the failure of many antibiotics to achieve sterility of bone after 4 weeks of therapy, except for clindamycin for S. aureus [41] and fluoroquinolones for Pseudomonas aeruginosa osteomyelitis [42]. Higher sterilization rates were achieved when rifampin was added to other active agents to treat S. aureus osteomyelitis even in the absence of prosthesis/hardware [43]. These experiments in animals have changed the way osteomyelitis is treated. Additional lessons included the discordant results of in vitro testing for synergy by adding rifampin and in vivo success, and the lack of correlation between bone penetration of antimicrobials and clinical efficacy [44].
Endocarditis
In 1912, Rosenow [45] claimed that he had produced endocarditis in rabbits using “exceedingly large” inocula of viridans streptococci. Grown in vitro, these organisms produce small clumps of bacteria, and Rosenow hypothesized that valves were infected by bacterial emboli. But the hemorrhagic lesions he observed were not consistent with the vegetations of endocarditis, and the bacterial inoculum was so large that most of his animals died within 24 h, presumably from cytokine storm. Nonetheless, a prevailing opinion [46] through the 1960s was that endocarditis resulted from emboli, not from adherence of bacteria to valve surfaces. Highman et al., [47] perforated the right coronary cusp of the aortic valves of 15 dogs and, at varying intervals thereafter, injected Staphylococcus aureus or viridans streptococci intravenously 4 times to 5 times weekly for 1 weeks to 3 weeks. All dogs developed typical lesions of endocarditis. Work of the heart and, later [48], turbulent blood flow were offered as explanations; no mention was made of bacterial adherence to damaged valvular tissue.
In 1970, Garrison and Freedman [49] developed a rabbit model of endocarditis, using intravenous catheters prefilled with a suspension containing 100 colony forming units (cfu)/ml of S. Aureus that traversed the tricuspid valve; endocarditis regularly resulted. Importantly, however, control rabbits, in which sterile catheters were placed also, developed valvular lesions. With remarkable prescience, Garrison and Freedman suggested that, should bacteria enter the bloodstream, platelet adhesion and serum factors would facilitate bacterial adherence, causing bacterial endocarditis. The following year saw the first report of bacterial endocarditis associated with a permanent transvenous pacemaker [50].
Freedman’s model was used to compare antibiotic prophylactic regimens against bacterial endocarditis caused by viridans streptococci [51] and to study antibiotic synergy in treating streptococcal and staphylococcal endocarditis, showing a good correlation between in vitro killing by various antibiotics singly or together and sterilization of cardiac vegetations [52-54] (see below, antibiotic synergy). This animal model elucidated several other important principles of infectious diseases:
(1) The role of serum factors such as fibronectin and fibrin in bacterial adherence to damaged valves [54];
(2) The absence of synergy between penicillin and an aminoglycoside in treating endocarditis due to highly aminoglycoside-resistant streptococci [55];
(3) The emergence of quinolone resistance during treatment of staphylococcal endocarditis with ciprofloxacin alone [56];
(4) Potential benefit of using rifampin [57], rifampin and a quinolone [58] or rifampin and vancomycin [59] in treating endocarditis; and
(5) The use of two cell wall-active agents (ampicillin and imipenem) in treating endocarditis due to multidrug resistant Enterococcus Faecium [60].
Intra-abdominal Infection
Despite numerous attempts to develop an experimental model of intraabdominal infection, none provided consistent results until Weinstein et al., [61] placed pooled, filtered colonic contents of meat-fed rats (colonic contents of rats on grain diets do not have the requisite diversity of anaerobic bacteria) into gelatin capsules together with barium sulfate and implanted them into the peritoneal cavity of rats. Most animals died within 72 h of bacteremia due to coliform organisms. Those that survived developed intraabdominal abscesses in which the predominant flora were Bacteroides [61, 62], a sequence of events replicating those, that follow perforation of the colon in humans. The model was then used to show that treatment with antibiotics directed against E. Coli (gentamicin) and Bacteroides (clindamycin) was effective [63]; these two drugs remained a staple in surgical practice for more than two decades. Bartlett et al used this model to compare 29 different antibiotic regimens in a remarkable study [64] that brought attention to the role for metronidazole or clindamycin together with agents effective against E. Coli in treating intraabdominal infection due to perforated bowel, .
Toxin-Mediated Disease
In the 1970s, a highly morbid “antibiotic-associated Pseudomembranous Colitis” was correlated with the use of clindamycin [65, 66]. Experiments in which clindamycin was administered to hamsters resulted in a similar enterocolitis and showed that a clindamycin-resistant, toxin-producing strain of Clostridium was the etiologic agent [67]. It was subsequently shown that:
(1) Broth cultures from the stool of patients with this disease frequently grew clostridia strains;
(2) Fecal transfer from diseased patients to hamsters caused enterocolitis; and
(3) Entercolitis in hamsters could be abated by concurrent use of gas gangrene antitoxin, implicating the role of toxin in this infection [68]. Experiments in hamsters finally identified toxin-producing Clostridium difficile as the causative agent, leading to development of the cell cytotoxicity assay as a diagnostic tool and to the use of oral vancomycin as a therapeutic option [69].
Toxin production is also central to the pathogenesis of Streptococcal Gangrene. After direct injection of > 103 Streptococcus Pyogenes into the muscle of mice, Eagle [70] found that organisms proliferated rapidly causing an infection that closely resembled streptococcal myositis in humans. When cfu in muscle exceeded 5 x 106, penicillin was no longer curative. Eagle’s explanation was that beta-lactam antibiotics only act on replicating bacteria. But organisms that no longer replicate may continue to produce toxins, and these toxins cause necrotizing fasciitis. Stevens et al., [71] first showed in vitro that clindamycin, but not penicillin, suppressed toxin production by Clostridium perfringens. They then reported that, after intramuscular challenge of mice with > 109 C. Perfringens, antibiotics that suppressed protein production, such as clindamycin and rifampin enhanced survival, whereas penicillin or cefoxitin did not [72]. In fact, in the mouse model, clindamycin was more effective than penicillin in treating streptococcal myositis [73]. These experiments showed that, rather than persistence of bacteria, toxin production caused disease, leading to the present practice of treating severe streptococcal infections of skin and soft tissue with antibiotics possessing anti-toxin activity such as clindamycin and, in more recent years, linezolid.
The pathophysiology of an uncommon disease, scalded skin syndrome, was elucidated entirely through animal experiments. This disease was identified in 1878 and attributed to an organism now presumed to be S. Aureus in 1891 [74], but it received little attention until 1956 when Lyell [75] described toxic epidermal necrolysis. Melish and Glasgow [76] reproduced it by infecting newborn mice with phage group 2 strains of S. Aureus and subsequently isolated a toxin (only from S. aureus of this phage group) that reproduced the syndrome in newborn mice [77].
Urinary Tract Infection
The pathogenesis of pyelonephritis was elucidated in animal experiments that began at the end of the 19th century. In 1921, Lepper [78] summarized earlier studies showing that: (1) introduction of coliform bacteria into the bladder of rabbits followed by urethral obstruction regularly caused cystitis; and (2) Coliform bacteria injected intravenously could be isolated from the kidneys. When she inoculated 108 Coliform bacteria intravenously during transient obstruction of one ureter, characteristic lesions of pyelonephritis appeared in the obstructed kidney but not in the nonobstructed one, findings that were later confirmed by Brumfitt and Heptinstall [79]. These experiments clarified the different pathogeneses of ascending and hematogenous infection of the kidneys, emphasizing the role of obstruction in both scenarios.
Animal studies showed that S. Aureus and Proteus mirabilis behaved differently from coliforms. When S. Aureus was injected intravenously into rabbits [80] or mice [81], organisms initially seemed to be cleared but, within 10 d, abscesses appeared in the kidneys in the absence of obstruction to urinary flow. These experiments led to the clinical observation that the finding of S. aureus in the urine in the absence of instrumentation should raise concern for prior or concurrent bacteremia [82]. Griffith and Musher [83] showed that introduction of a foreign body and P. Mirabilis into the bladder of rats caused infection with formation of struvite stones; concurrent administration of a urease inhibitor prevented stone formation and greatly reduced the extent of kidney involvement [84] again documenting that bacterial products, not simply persistence of live bacteria, produce disease.
Norden et al., [85] reported that attachment of E. Coli to uroepithelial lining cells was followed by loss of recoverability of the bacteria. Subsequent electronic microscopic studies in infected rats [86] showed, in fact, that uroepithelial cells actually phagocytose bacteria and that these cells are then shed into the urine. These findings explain the how incomplete emptying of the bladder increases the risk for infection.
Neutropenia and Synergistic Effect of Antibiotics
With the advent of novel chemotherapies in the 1960s, neutropenia and associated infections due to Gram negative bacilli became increasingly prevalent. Because of the clinical variability among patients, direct studies in humans were difficult, and an in vitro “checkerboard” technique to examine synergy in vitro yielded inconsistent results with poor applicability to infected patients [59]. In an important advance, Andriole [87] demonstrated synergy between carbenicillin and gentamicin in rats after lethal challenge with Pseudomonas even when in vitro synergy could not be demonstrated. Using neutropenic rats, Lumish and Norden [88] studied varying doses and dosing intervals of aminoglycosides, penicillins and cephalosporins singly and in combination after intraperitoneal, intrabronchial or intramuscular inoculation with Klebsiella or Pseudomonas [89, 90] ; synergy between beta-lactams and aminoglycosides was consistently demonstrated.
The so-called post-antibiotic effect of aminoglycosides [17], now more clearly understood to be related to ribosomal binding at peak concentrations, was documented in this same model. More recently, meropenem/ vaborbactam was found to be modestly useful in treating neutropenic mice infected with carbapenemase-producing Enterobacteriaceae [91], organisms that have been notoriously difficult to eradicate and for which usually available in vitro testing yields inconsistent results. This same neutropenic mouse model has also been used to optimize dosing regimens for new antibiotics such as solithromycin that may have unusual volumes of distribution and/or pharmacodynamics [92].
Spanning airborne transmission of infection, immunologic response to pathogens, discovery of disease mechanisms, pharmacologic principles of antimicrobial therapy and impact of a foreign body on establishment and persistence of infection, animal studies have contributed immensely to the field of infectious diseases, and provided a foundational understanding for our approach to many classic infectious disease syndromes we encounter today including endocarditis, pyelonephritis, intraabdominal infection and osteomyelitis. In this article, we hope to have inspired the reader to read and reflect upon this important historical body of literature.
Not applicable.
No conflict.
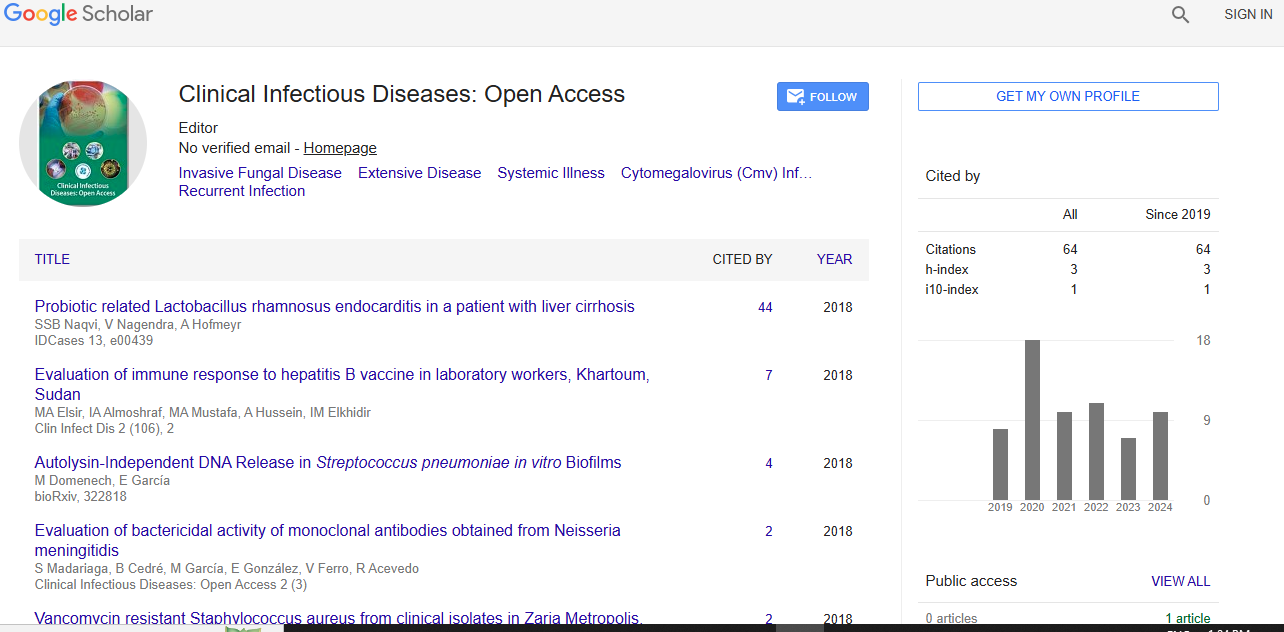
Clinical Infectious Diseases: Open Access received 1149 citations as per Google Scholar report