Review Article - (2023) Volume 13, Issue 2
Received: 10-Jul-2023, Manuscript No. JBL-23-105357;
Editor assigned: 14-Jul-2023, Pre QC No. JBL-23-105357;
Reviewed: 28-Jul-2023, QC No. JBL-23-105357;
Revised: 04-Aug-2023, Manuscript No. JBL-23-105357;
Published:
11-Aug-2023
, DOI: 10.37421/ 2165-7831.2023.13.303
Citation: Misra, Srimanta Chandra, Gaetano Lucania,
Valentina Guarino and Alberto Santagostino. " The New Era for
Advancements in Peripheral T-Cell Lymphoma: A Review" J Blood Lymph 13(2023):303.
Copyright: © 2023 Misra SC, et al. This is an open-access article distributed under the terms of the creative commons attribution license which permits unrestricted
use, distribution and reproduction in any medium, provided the original author and source are credited.
Peripheral T-Cell Lymphomas (PTCL) are an uncommon and heterogeneous group of disorders arising from the innate and adaptive immune system. This review deals with the basis and major revisions to the current PTCL classification. Each entity in the 2016 classification has been reviewed on the basis of cell origin, genetic landscape and recent therapeutic options. The objective of this study was to conduct a review of the normal immune system, signalling pathways and tumor microenvironment in order to understand the heterogeneity of certain entities as well as to uncover the potential therapeutics. A brief evaluation of a normal immune system, implication of the JAK-STAT pathway and tumor microenvironment was performed to explain the heterogeneity of PTCL. Attempts were also made to optimize current standard and personalized management approaches. Fulfilling the current unmet needs in PTCL require optimization of the intensity and number of courses of chemotherapy in first-line treatment, choosing the right strategy of intensification such as ASCT versus improved HSCT and lastly, tailoring the salvage treatment within the currently available options including HSCT, chemo-immunotherapy and targeted therapy. Further knowledge would pave a better future to better manage PTCL.
Peripheral t-cell lymphoma • PTCL • Tumor microenvironment • Signaling pathways
AITL: Angioimmunoblastic T-cell Lymphoma; ASCT: Autologous stem cell transplant; ATLL: Anaplastic Large Cell Lymphoma; COO: Cell of Origin; ETTL: Enteropathy-Type T-Cell Lymphoma ; HSTCL: Hepatosplenic T-Cell Lymphoma; MHC: Major Histocompatibility Complex; MEITL: Monomorphic Epitheliotropic Intestinal T-Cell Lymphoma; NHL: Non-Hodgkin’s Lymphomas; ORR: Overall Recovery Rate; PC-ALCL: Primary Cutaneous Anaplastic Large Cell Lymphoma; PTCL: Peripheral T-cell Lymphomas; PTCLNOS: Peripheral T-Cell Lymphoma Not Otherwise Specified; SCT: Stem Cell Transplantation; SPLTCL: Subcutaneous Panniculitis-Like T-Cell Lymphoma; TCR: T-Cell Receptor; TME: Tumor Microenvironment
As a whole, Peripheral T-Cell Lymphoma (PTCL) represents approximately 6% to 10% of all Non-Hodgkin’s Lymphomas (NHL) arising from the different maturation stages of T and NK cells. [1]
Both of these cells are responsible for adaptive and innate immunity and in regard to their origin, clinical behavior and microenvironment, they are quite heterogeneous. Most of the forms of NHL carry a dismal prognosis despite conventional chemotherapy and Stem Cell Transplantation (SCT) efforts and constitute a serious unfulfilled need since only a few of them undergo an indolent course. For emerging perspective therapeutics, a brief description of the T-cell ontogeny and the JAK-STAT signaling pathway involved in the process is essential.
The objective of this study was to conduct a review of the normal immune system, signaling pathways and tumor microenvironment in order to understand the heterogeneity of certain entities as well as uncover potential therapeutics.
The review was performed by searching for articles according to the MECIR guidelines and with keywords such as PTCL including each individual entity of PTCL as a keyword. A second search was made using the keywords “signaling pathways” and “tumor microenvironment” in relation to PTCL. Due preference was given to the most recent pub` lications.
The international PTCL study performed in 2008, (which was before the 2016 classification revision) found that Peripheral T-cell lymphoma not otherwise specified (PTCL-NOS) constituted most cases (25.9%) followed by Angioimmunoblastic T-cell Lymphoma (AITL) (18.5%), Anaplastic Large Cell Lymphoma (ALCL)(12.5%), Natural Killer T-Cell Lymphoma, (NKTCL), Adult T-Cell Leukemia/ Lymphoma (ATLL), Enteropathy-type T-Cell Lymphoma (ETTL), Primary Cutaneous Anaplastic Large Cell Lymphoma (PC-ALCL), Hepatosplenic T-cell Lymphoma (HSTCL), Subcutaneous Panniculitis-like T-cell Lymphoma (SPLTCL) as well as other rare subtypes (Table 1 and Figure 1) [1].

Figure 1. Relative frequency of different types of PTCL.
The pitfalls of cell of origin based classification
Although the ontogeny of lymphoid cells has been better understood in recent years with the Cell of Origin (COO) based classification, the normal counterpart of neoplastic cells cannot be the sole basis of the division. This is because of the immunophenotypic heterogeneity of some of the entities that are found in HSTCL which are derived from either α/β or γ/δ T cells [2].
The tumor microenvironment
The COO based classification generally disregards the Tumor Microenvironment (TME) which could not only endorse a diagnostic and prognostic significance but could also be of therapeutic interest. TME around the Hodgkin Reed Sternberg Cells (HRS Cells) are well studied which are rich in inflammatory and immune infiltrates. TME consists of T Infiltrating Lymphocytes (TILs), Macrophages (M1 and M2), eosinophil and mast cells. Most TILs are CD8 T-cells engaging MHC Class I on tumor cells via TCR (T Cell Receptor). The other cells are CD4+ cells including T Follicular Helper Cells (TFH), T-regulatory (Treg) and Th17 cells in addition to those of Bcell lineages (including B cell blast and plasma cells). The AITL has an immunological TME consisting of eosinophil, Bcell blast, plasma cells and macrophages [3], whereas PTCL-NOS is characterized by an abundance of mast cells. TH17 cells and the number of Treg are similar in both entities.
Mast cells strongly expressing IL6 might affect a pro-inflammatory environment and mediate a balance between immune regulation and autoimmunity in AITL [4]. Takesi, et al. [5] analyzed the gene expression signatures of tumors and tumor infiltrating immune cells including B-cell, Dendritic Cell (DC), mast, neutrophil, eosinophil, macrophage, natural killer (NK) as well as T-cell subtypes (Th1, Th2, Th17, TFH γ δ T-cell (Tgd), memory T-cell (Tm) and CD8+ T cell) in PTCL-NOS. They found that the cases with B-cell and DC signatures (BD group) showed favorable clinical outcomes, whereas the non-BD group had poorer prognoses. Half of the non-BD cases carry a macrophage signature. The macrophages harbor immune check point molecules such as PD ligands [6].
The immune system
The central lymphoid tissue includes bone marrow and the thymus, whereas the spleen, lymph node mucosa, peripheral blood and the skin constitute the peripheral lymphoid tissue. Both parts orchestrate the innate and adaptive immune system in a coordinated manner andhe innate immune system includes NK cells, γδ T cells and NKlike T cells that constitute a non-specific, non-memorized primary immune response.
The adaptive immune system is made up of αβ T cells which leave the thymus and, upon antigenic stimulation, differentiate to CD4+ and CD8+ effector and memory cells. In fact, CD8+ T cells are composed of a α and β chain recognize MHC class I molecules and are responsible for the production of at least two distinct families of cytotoxic granules (perforin and granzyme-like) NK cells. This partly explains the incidence of hemophagocytic lympho-histiocytosis (HLH) in some PTCL. CD4+ T cells recognize the MHC Class II molecules on Antigen-Presenting Cells (APC). Th17 cells are subsets of CD4+ cells which are linked to diverse inflammatory conditions such as arthritis and Inflammatory Bowel Disease (IBD). Another subset is the Treg Cells (CD4+, CD25+ and Foxp3) which are responsible for homeostasis and tolerance of the immune system. The CD4 subtype of current interest concerns TFH cells which are located in the Secondary Lymphoid Organs (SLO) such as the tonsils, spleen and lymph nodes. In the T-cell zone of SLO, CD4 T cells are recognized via the TCR protein present on the MHC molecule of Dendritic Cells (DC) and thus are committed to TFH lineage expressing Bcl6. At the border of the T and B cell zones, they acquire CXCR5, ICOS PD1 and subsequently migrate to the B cell zone. A fully differentiated TFH expresses a high level of CXCR5 and ICOS PD 1, which helps the formation of Germinal Center (GC) and prompts B cells to produce antibodies.
Concerning the innate immune system, the γδ T cells are activated in an MHC independent manner and often present a tissue-specific localization in epithelial and mucosal tissues. The NK cells receive inhibitory signals from normal cells expressing MHC Class 1 molecules. Tumor and virally infected cells are ideal prey for NK cells since they ultimately down regulate MHC Class 1 molecules. Being the largest organ of the body, the skin serves as an epithelial barrier consisting of one million T cells containing 1% to 10% of γδ T cells and the remaining population being αβ T cells per square centimeter. Vitamin D induces CCR 10 expression on T cells which is responsible for skin homing of T cells [7].
The Intraepithelial T Lymphocytes (TIELs) maintain the epithelial barrier in the gut, which is the largest T cell compartment of the body, and has a ratio of I T IEL per 10 epithelial cells [8]. T IEL homing and retention is accomplished by gut homing molecules such as integrin and CCR 9. Retinoic acid is a key inducer of gut homing molecules. CXCR3 is expressed on activated T cells including CD8+ IEL [9]. Lymphomas arising from the innate immune system are predominantly extra snodal, whereas those arising from the adaptive immune system mostly affect adults and are mainly nodal in origin (Figure 2).

Figure 2. T and NK Cell differentiation and maturation (courtesy of Lucania L).
The JAK-STAT pathway
The JAK-STAT signaling pathway plays a crucial role in T-cell survival, proliferation and differentiation and involves three major proteins: i) cell surface receptors, ii) Janus Kinase, and iii) Signal Transducer and Activators of Transcription (STATs). There are four JAK proteins (JAK1, JAK2, JAK3 and TYK) and seven STAT proteins (STAT 1, STAT 2, STAT 3, STAT 4, STAT 5A, STAT 5B and STAT 6). The binding of various ligands (mainly cytokines) to cell surface receptors leads to phosphorylation of JAKs, which, in turn, phosphorylates STATs to form a dimer that enters the nucleus where it binds to DNA causing transcription of the target gene. Cytokines that signal through receptors containing the common γ Chain (γC) subunit are pivotal for immune hemostasis. The γC dependent cytokines are IL 2, IL 4, IL 7, IL 9, IL 15 and IL 21. Disorders involving γC/JAK-STAT are identified in most T-cell leukemia-lymphoma [10,11].
What have we learned from the 2016 revision?
The potential changes in PTCL that prompted the 2016 revision mainly included the entities of PTCL-NOS, ALK-negative ALCL, Tcell lymphomas of the GI tract, cutaneous T-cell lymphomas and Epstein-BarrVirus (EBV)-positive NK/T Cell neoplasm. The T-cell lymphomas of TFH origin not meeting the criteria of AITL are grouped under Nodal T-cell Lymphoma with TFH phenotypes being displaced from PTCL-NOS. Follicular T-cell lymphoma is a new provisional entity with a TFH phenotype and follicular involvement patterns. The ALK-negative ALCL has been upgraded to a definite entity and the genetic subgroup involving DUSP-IRF4 has been recognized with a favorable outcome. Moreover, Breast Implant Associated Lymphoma ALCL (BIA ALCL) has become a provisional entity. The nomenclature “EATL” is now restricted to what was previously known as EATL 1 (associated with Celiac Disease (CD)) and the previous EATL II (non-associated with CD) is now known as MEITL. Additionally, Indolent T Lymphoproliferative Disorder (TLPD) of the GI Tract has become a provisional entity. Primary cutaneous CD4-positive small/medium TLPD is no longer considered as a lymphoma because of its indolent behavior. Primary cutaneous acral CD8-positive T-cell lymphoma is also a provisional entity. Because of the aggressive behavior, Childhood Systemic EBV-positive T-cell lymphoma has ceased to be considered as a lymphoproliferative disorder [12,13].
T-PLL is a rare, mature, post thymic T-cell tumor which shows a refractory behavior towards CHOP (Cyclophosphamide, Hydroxydaunorubicin, Oncoven and Prednisone/Prednisolone) therapy. The current treatment of alemtuzumab followed by Allo HSCT produces a 4-year PFS of 30% and 4-year OS of 42%. However, only 30% to 50% of patients are eligible for HSCT [14]. Mutations in JAK3 and STAT 5B as well as in JAK1 have been documented in a few cases. It is probable that CAR T could target the constant region of the TCR B chain. This concept was recently hypothesized by Wahnschaffe and Herling [15]. CCR 7 is expressed in most of T-PLL indicating that the anti-CCR monoclonal antibody needs further trials.
T-cell LGL arises from the CD8+ alpha beta cell which has an indolent course. A constitutive activation of STAT 3 accompanied by STAT 3 gain of function is found in 40% to 70% of all cases [16]. Immunosuppressors are the first-line therapy followed mostly by purine analogs as a second-line therapy. Rituximab is used in patients with Rheumatoid Arthritis and LGL. Even splenectomy in refractory cytopenia in T-LGL confers a sustained response in 56% of cases [17]. The chronic lymphoproliferative disorder of NK cells arises from mature NK cells and mostly involves the STAT 3 and JAK3 mutations [18]. It is an indolent disease treated with singleagent immunosuppressor.
Aggressive NK cell leukemia arises from activated NK cells. Although rare, it carries a grave prognosis and is often present with DIC and hemophagocytic syndrome. EBV-positive cases are positive for EBER and negative for LMP suggesting a Type I latency [19]. Activation mutations in STAT 5B, STAT 3, JAK3 are found along and within the signaling pathways such as AKT and NF kappa B. Although there is no consensus on management, L asparaginasebased treatment followed by Allo SCT is the treatment of choice [20].
Childhood EBV T cell lymphoma arises from a cytotoxic CD8+ T cell or activated CD4+ T cell. LMP1 prohibits apoptosis by upregulation of Bcl2 and activation of the NFKP pathway. R CHOP remains the treatment of choice, whereas Allo SCT is a limited option because of its toxicity [21]. Hydroa vacciniforme– like lymphoproliferative disorder occurs mainly in Latin American and Asian countries and emanates from chronic active EBV infection to affect skin-homing T or NK cells. The management of this disorder depends on its stage and consists of either immunosuppressive therapy/immunomodulators alone or systemic NHL therapy [22].
Adult T-cell leukemia/lymphoma is caused by HTLV-1 (most probably originating from regulatory T cell (Treg) cells (CD4+, CD25+, FoxP3)). The majority of patients with ATLL harbor mutations in TCR/NFKB activation pathways.
There are four different types of presentations: acute, lymphomatous, chronic and smoldering. ATLL cells show high expression of chemokine receptors such as CCR4 and CCR7 which are involved in the migration and infiltration of lymphoid organs and skin. The expression of PD-L1 in ATLL cells has been correlated with poor prognoses. Dose-adjusted EPOCH with maintenance with Ziduvudine yields 58% of ORR, while dose-adjusted EPOCH with Raltegravir and Bortezomib has an ORR of 67% [23]. Mogomulizumab (an anti-CCR antibody) is approved in Japan for relapsed/refractory patients [23]. In a phase II study, Linolidamide had an ORR of 42% [24].
Extranodal NK-/T-cell lymphoma (nasal type) originates from NK or γδ T cells which both express CD 56. EBV plays an etiological role. The growth is supported by activation of the JAK/STAT pathway (STAT 3 STAT 5B JAK3 and PTPRK) [25]. The mutation of the cell surface receptor is detected in more than half of the patients and p53 mutation is seen in 20 to 50% of patients. Nevertheless, the gene mutations related to epigenetic modification are found in NKTCL including genes associated with histone methylation (KMT2D), histone acetylation (EP300), histone deubiquitylation (ASXL3) and chromatin remodeling (ARID1A) [26].
Chemoradiotherapy is the main management strategy using L asparaginase-containing regimens such as SMILE (dexamethasone, methotrexate, ifosfamide, l-asparaginase, and etoposide). The anti- CCR antibody, Mogomulizumab and/or checkpoint inhibitors might be potential therapeutics in the future [27].
The most recent revision of the PTCL classification distinguishes Enteropathy-Associated T-cell Lymphoma (EATL) arising from de novo or refractory celiac disease (previously known as EATL 1) or from Monomorphic Epitheliotropic Intestinal T-cell Lymphoma (MEITL) which was previously known as EATL 2 [27,28]. The cell of origin is the small intestine cytotoxic intraepithelial TCRαβ+ T cell and also the TCRγδ+ T cell in some limited cases. JAK 1 and STAT 3 mutations have been described in refractory celiac disease. Due to poor prognosis and an aggressive course, combination chemotherapy with ASCT is the treatment of choice [29].
MEITL is a provisional entity originating from γδ T cells or, more rarely, from αβ+ intraepithelial T cells which are mostly CD30+ negative negative and frequently involves the jejunum. Combination chemotherapy with ASCT is the treatment of choice; however, CHOP is insufficient to induce remission [30]. Recurrent alterations in SETD2 are characteristics of MEITL and are detected in >90% of patients [31]. Additional mutations involving epigenetic regulators like EZH2 are also found. The important driver mutations are STAT 5B and JAK3, and the STAT 5 inhibitor, Pimozide, merits a clinical trial in refractory situations [32].
The Indolent T-cell lymphoproliferative disorder of the GI tract originates from mature peripheral CD4+/CD8+ T cells. The STAT 3 mutation is absent and EBER is negative. This has an indolent course and radiotherapy is the preferred option [33,34].
Hepatosplenic T-cell Lymphoma is a rare neoplasm arising mainly from non-activated cytotoxic γδ T cells and, in a few cases, from α/β T cells. They express cytotoxic markers such as T1A1 and granzyme M. The symptoms include hepatosplenomegaly and cytopenia, which are sometimes associated with hemophagocytic syndrome. In addition to the frequent mutations in chromatin modifier genes, Next-Generation Sequencing (NGS) reveals genetic alteration in the JAK/STAT pathway (STAT 3 and STAT 5B), epigenetic regulation and in the P3K signaling pathway. Two single-institution studies reported poor results with CHOP or CHOP-like regimens [35,36]. Following an initial response, SCT is promising. Furthermore, ifosfamide/cytarabinebased regimens are used for salvage therapy if CHOP-like regimens are used as first-line treatments. Alemtuzumab may have efficacy in relapses or in association with cladribine. To date, the best approach seems to be a Hyper CVAD-like regimen followed by SCT [37]. Subcutaneous Panniculitis-like T-cell Lymphoma is a rare primary cutaneous T-cell lymphoma of mature cytotoxic CD8 T cells. Immunosuppressive agents are considered as first-line treatments [38]. There are recent reports about lenalidomide as well as chidamide that offer long-term remission [39].
Mycosis Fungoides (MF) and Sézary Syndrome (SS) constitute 75% of the cases of Cutaneous T-cell Lymphoma (CTCL). MF is a low-grade cutaneous lymphoma arising from skin-homing CD4+ T cells, whereas SS arises from skin-homing central memory T cells. The recurrently affected genes in MF/SS include TP 53, STAT 5B and DNMT3A. In the early stage, skin directed immunosuppressive therapy is employed [40] and with the aggressive form, polychemotherapy is used [41]. Recently, targeted therapies have shown promising results. Brentuximab Vedotin (BV) has been approved for CD30+ cutaneous T-cell lymphoma. Magamulizumab has shown more significant ORR and PFS than Vorinostat [42]. Alemtuzumab shows an ORR of 55% when used in the erythematous form of MF/SS [43]. Nonmyeloablative HSCT for CTCL has 46% OS at five years [44].
Primary cutaneous CD30+ T-cell lymphoproliferative disorders issuing from activated skin-homing T cells is the second most common cutaneous lymphoma and consists of subtypes such as Lymphomatoid papulosis, Primary cutaneous anaplastic large cell lymphoma and borderline lesions. The former has an excellent prognosis, whereas the latter has a poorer one. The anti CD30, Brentuximab Vedotin (BV), is a therapeutic target [45-47].
Primary cutaneous γδ T-cell lymphoma develops from mature and activated γδ T-cells with cytotoxic phenotypes. Daniels, et al. identified 20 driver mutations including STAT 3 STAT 5B and other mutations in MAPK signaling. K-RAS is most frequently mutated [48]. Due to the resistance towards chemotherapy and radiotherapy, no standard treatment is defined. However, the CHOEP and BV CHP merit mention. There are case reports concerning the activity of BV Gemcitabine [49].
Primary cutaneous CD8+aggressive epidermotropic cytotoxic Tcell lymphoma arises from skin-homing CD8+ T cells. The conventional multi-agent chemotherapy treatment has an unsatisfactory patient’s outcome. In these cases, HSCT is the treatment of choice [50]. Primary cutaneous acral CD8+ T-cell lymphoma is a slow-growing lesion amenable to local excision or topical steroids. Interferon and PUVA therapy is used for multifocal diseases [51]. Primary cutaneous CD4+ small/medium T-cell lymphoproliferative disorder has an indolent behavior with uncertain malignant potential. Arising from skin homing CD4+ cells with TFH features, excision or local radiotherapy are almost always curative [52].
Peripheral T-cell lymphoma NOS represents highly heterogeneous post thymic T-cell neoplasm and accounts for 35% of PTCL cases in the western world. It has a clonal TCR arrangement and an aberrant T-cell phenotype with a loss of CD5+ and CD7+. It also has a clonal TCell Receptor (TCR) gene rearrangement [53]. The Anthracyclinebased combination regimen followed by ASCT is the treatment of choice [54]. The molecular landscape of PTCL-NOS reveals frequent mutations in the different epigenetic regulators and the genes involved in NF-κB and TCR signaling pathways. Amador, et al. proposed an algorithm for molecular sub-classification of PTCL [54,55]. GATA 3 and PTCL TBX 21 with distinct oncogenic pathways and prognoses represent 33% and 49% of PTCL-NOS, respectively. GATA 3 is a regulator of TH 2 differentiation and has an outcome inferior to that of PTCL TBX 21 subtypes. TBX 21 is a master regulator of TH1 cells.
Angioimmunoblastic T-cell Lymphoma (AITL) is the second most common type of PTCL stemming from T Follicular Helper Cells (TFH). It has drawn recent attention because of its TFH markers and recurrent mutations that affect epigenetic regulators such as TET2, DNMT3A, and IDH2. The 2016 WHO revision requires detection of two or three out of seven antigens (CD10, BCL6, PD1, CXCL13, CXCR5, ICOS, and SAP) to ascertain the origin of TFH. The understanding of TFH function in the formation and reaction in GC explains the immunological abnormalities encountered in AITL. In addition, there are recurrent mutations in TET2, DNMT3A, IDH2 and RHOA in 80%, 20%-40%, 20%-30% and 50% to 70% of cases, respectively [56]. This heterozygous, missense RHOA G17V mutation, which is highly specific to AITL, can coexist with mutations of epigenetic regulators and carries a relatively worse prognosis. The lymphomagenesis of AITL downstream of AITL depends upon the ICOS/PI3K/mTOR signaling pathway. The VAV1 mutation is found in 1% of PTCL cases and is mutually exclusive with RHOA mutation. A recurrent activating mutation in CD28+, a costimulatory molecule of T Cell Receptor (TCR), has been described in AITL [57].
The findings of de Leval, et al. strongly contributed to the fact that TFH cells are the normal counterpart of AITL, and suggest that the AITL spectrum may be wider than suspected, since a subset of CD30(-) PTCLs-U (peripheral T-cell lymphoma unspecified) may derive from or can be related to AITL [58]. However, the CHOP (cyclophosphamide, doxorubicin, vincristine, and regimen and consideration for autologous Stem-Cell Transplant (SCT) remains the current frontline approach [59].
In AITL, EBV+ B cells are found in the majority of cases that might undergo clonal expansion of B cells giving rise to DLBCL [60]. In AITL, the adjunction of Rituximab in standard CHOP-based therapy yields a response rate of 80% as found in one Phase 2 trial [61]. Because of mutations in epigenetic regulators, hypomethylators such as oral Azacytidine and Romidepsin do hold some promise [62]. The response to PD1/PDL 1 inhibitors and the multikinase inhibitor, Dasatinib, merits future trials [63]
Follicular T-cell lymphoma and Nodal peripheral T-cell lymphoma with TFH phenotype share the same origin and gene expression as AITL with the difference being that the former presents with the absence of proliferative vessels or EBV positive immune-blasts and the latter shows diffuse infiltration without polymorphic inflammatory background [64].
Anaplastic large-cell lymphoma could be either ALK+ or ALK-. ALK-positive cases have a better prognosis. ALCL arises from activated cytotoxic T cells. In ALK-negative cases, the molecular events such as DUSP22 and TP 63 affect survival [65]. The former carries a favorable prognosis, whereas TP 63 has an unfavorable one. Frequent activating mutations in JAK 1 and STAT 3 have been identified in ALK-negative ALCL.
ALK translocations results in the activation of JAK/STAT and in PI3K signaling pathways. The activation of STAT 3 has been shown to be a requirement for the maintenance of the transformed phenotype in ALK+ALCL. Therefore, STAT 3 seems to be the common mechanism of malignant transformation, irrespective of any ALK status [66].
Multiagent CHOP-like chemotherapy remains a standard of care for newly diagnosed ALCL patients treated with curative intent and provide a chance of cure for the majority of ALK-positive ALCL patients, and at least half of the ALK-negative ALCL patients [58]. Brentuximab/Vedotin could be an option in refractory patients or as first-line strategies for frail patients [67]. BV-CHP needs to be assessed as a front-line treatment. Successful treatment with Crizotinib (an ALK inhibitor approved for Non-Small Cell Lung Cancer) has been reported [68].
Lastly, the provisional category of Breast Implant-associated Anaplastic Large-cell Lymphoma (BIA ALCL), which has for a longtime been considered as a subtype of ALCL, is associated with prolonged exposure to breast implant(s). The frequency varies from 1 in 500,000 to 1 in 3 million even though there are over 10 million patients worldwide with breast implants. This ALK-negative CD30- positive ALCL mostly presents itself in the form of localized disease and surgical excision is crucial. ALCL-based management is mandated in advanced disease cases [69].
Finally, the inhibition of STAT 5 could be a promising approach for PTCL. A better understanding of Pimozide and related molecules might pave the way for inducing apoptosis in STAT 5 induced PTCL. Fulfilling the current unmet needs in PTCL require optimization of the intensity and number of courses of chemotherapy in first-line treatment, choosing the right strategy of intensification such as ASCT versus improved HSCT and lastly, tailoring the salvage treatment within the currently available options including HSCT, chemo-immunotherapy and targeted therapy. Further knowledge would pave a better future of management of PTCL.
The authors would like to thank Dr. Stephane Sanchez, the clinical research director, for his technical help and Lucio Lucania for the figures. The authors would also like to thank AcaciaTools for their editing and writing services.
The authors declare no conflict of interest.
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
[Crossref][Google Scholar] [PubMed]
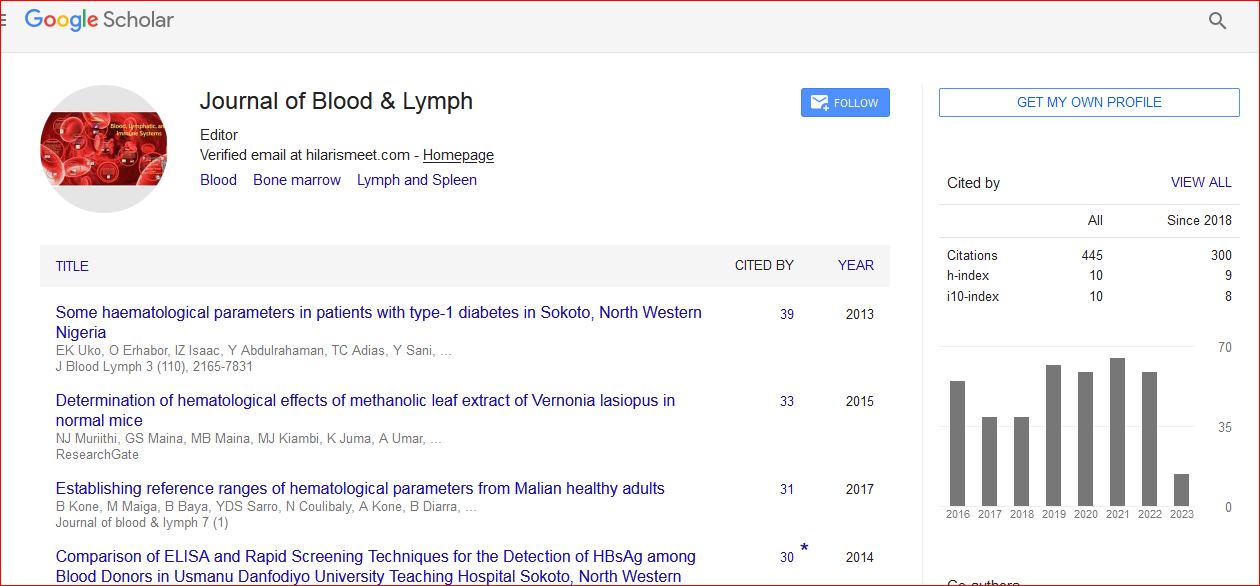
Journal of Blood & Lymph received 443 citations as per Google Scholar report