Research Article - (2021) Volume 10, Issue 5
Received: 20-Aug-2021
Published:
10-Sep-2021
, DOI: 10.37421/2470-6965.2021.10.167
Citation: P Patowary, B Deka, D Bharali. "Tetrazole Moiety as
a Pharmacophore in Medicinal Chemistry: A Review." Malar Contr Elimination
10 (2021): 167.
Copyright: © 2021 B Deka, et al. This is an open-access article distributed under
the terms of the Creative Commons Attribution License, which permits unrestricted
use, distribution, and reproduction in any medium, provided the original author
and source are credited.
In present day scenario, tetrazole has gained increasing popularity due to its broad-spectrum of biological properties such as antitubercular, anticancer, antimalarial, antidiabetic, antifungal, anti-inflammatory activities etc. Tetrazole, beinga bioisostere of the carboxylic acid group can replace the carboxyl group in drugswhich can be utilized to increase the lipophilicity, bioavailability and to reduce theside effects of drugs. Many tetrazole containing compounds have already been used for the treatment of various diseases primarily due to their better pharmacokineticprofile and metabolic stability. Therefore, this heterocyclic tetrazole moiety having admirable biological, pharmaceutical and clinical applications can be considered as an important pharmacophore in the development of new drugs.The presentsurveyreviews the various approaches available for the synthesis of tetrazole derivatives along with the different biological activity of substituted tetrazole derivatives like antitubercular, anticancer, antimalarial, antidiabetic, and antifungal and anti-inflammatory. This survey will serve as a comprehensive database of the biological, pharmaceutical and clinical applications of tetrazole which will be beneficial in facilitating further research and development on the topic.
Tetrazole • Synthesis • Anti-inflammatory • Anti-diabetic • Anticancer • Antitubercular • Antimalarial • Antifungal activity
Tetrazole have attracted significant interest in the last few decades. These are the heterocyclic nucleus which contains a five membered ring of four nitrogen, one carbon and two hydrogen atoms. It is basically an aromatic aza-pyrrole nucleus, which may exist in two tautomeric forms 1 and 2. The molecular formula of tetrazole is CN4H2. The numbering system indicated in 1 and 2 is that used throughout the review. The structural unit, -C(N3) =N-, is described either as an azidoazomethine, an iminoazide, or an imidoylazide, all of which terms are used widely in the literature [1] (Figure 1).

Figure 1. The numbering system indicated in 1 and 2 structural unit, -C(N3) =N-, is described either as an azidoazomethine, an iminoazide, or an imidoylazide.
Replacement of one of the ring protons leads to three possible types of mono substituted tetrazoles with substituents at the 1, 2 or 5 positions respectively. Replacement of two ring protons leads to two possible types of disubstituted tetrazoles namely 1, 5 disubstituted and isomeric 2, 5- disubstituted derivatives [2].
Tetrazole is white to pale yellow crystalline solid which has a weak characteristicodour and soluble in water or alcohol. It is acidic in nature due to the presence of four nitrogen atoms[3].
The tetrazole functional group acts asa metabolically stable isostere for the carboxylic acid and this has been a primary driving force for continual research in the area [4].
The first tetrazole was prepared in 1885 by the Swedish chemist J.A Bladin at the University of Upsala during the course of an investigation of the reactions of dicyanophenylhydrazine, the condensation product of cyanogen and phenylhydrazine. Bladin observed that the action of nitrous acid on dicyanophenylhydrazine led to the formation of a compound C8H5N5, to which he described the formula (Figure 2).

Hydrolysis, followed by decarboxylation, produced a compound having the formula C7H6N4, C6H5CN4 structure unit remaining intact throughout these transformations. During his study of dicyanophenylhydrazine, Bladin has prepared numerous triazoles and the possibility of forming a nitrogen heterocycle with one more ring nitrogen was a logical extension of his interpretations. The following year Bladin proposed the name “tetrazole” for the new ring structure and in 1892 succeeded in preparing tetrazole itself by various series of reactions, starting with carboxylic acid produced from his phenylcyanotetrazole [5].
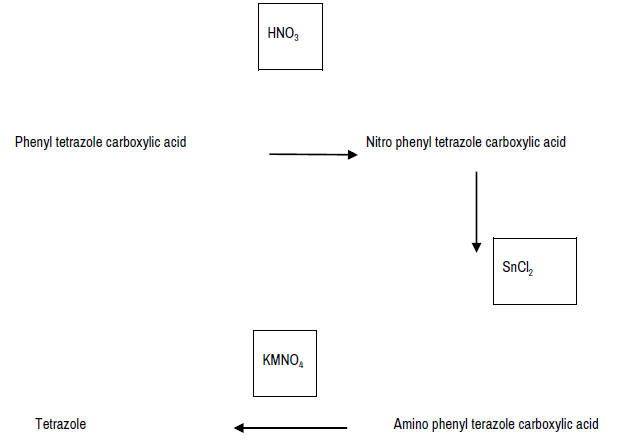
Among the family of nitrogen-containing heterocycles, tetrazole and its derivatives exhibit a large range of biological properties such as antibacterial, antifungal, anticancer, analgesic, anti-inflammatory, antidiabetic, antihyperlipidemic, anti- malarial and anti-tubercular activities. The unique features of tetrazole, biological significance and applications are discussed in this review. It is the structural component in many drugs, drug candidates, lead compounds and various biochemical reagents.
The introduction of the tetrazole ring into a molecule of an organic substrate quite often leads not only to an increase in the efficacy but also to an increase in the prolongation of drug action. As a rule, this is not accompanied by an increase in acute toxicity. It is no coincidence that the World Health Organization declared the tetrazole ring as an important descriptor in the methodology of the design of new drugs using the analogue based drug discovery (ABDD) method [6].
Being a bioisostere of the carboxylic acid group, it can replace carboxyl group in drug molecules to increase lipophilicity, bioavailability and reduce side effects so tetrazole derivatives occupy an important position in the discovery of new drugs [7].
Tetrazole in anticancer activity
Cancer is the second leading cause of death globally, accounting for an estimated 9.6million deaths, or one in six deaths, in 2018. Lung, prostate, colorectal, stomach and liver cancer are the most common types of cancer in men, while breast, colorectal, lung, cervical and thyroid cancer are the most common among women [8].
More than several hundred of drugs exist in the market with a very serious draw back as they are unable to differentiate between normal cells and cancerous cell types [7].
Cancer cells become simultaneously resistant to different structural types of chemo therapeutic agents due to the multidrug resistance. Hence, the development of efficient, selective and less toxic anticancer agents is necessary to overcome the multidrug resistance of cancer cells [9].Compared with the parent drugs, hybrid molecules have the potential to improve the efficacy, over come drug resistance and reduce the toxicity. Therefore, hybridization of tetrazole and pharmacophores with anticancer properties represents a promising strategy to develop novel anti-cancer candidate [9].
Chebolu Naga et al. synthesized some new tetrazole derivatives by Baylis– Hillman allyl amines and they were screened against selected human cancer cell linesof liver carcinoma (Hep-G2), lung adenocarcinoma (A-549), breast (MDA-MB-231), prostate carcinoma (DU-145), neuroblastoma (SKN- SH) origin. Sulforhodamine B(SRB) colometric assay method was used to evaluate the IC50.The compounds 3a,3c,3e and 3f displayed the highest in vitro anticancer activity (1.0–4.0 mM) on both liver carcinoma (Hep G2) and lung adenocarcinoma (A 549) cell lines. Compounds 3b, 3c, 3d and 3f displayed significant activity (4.0–7.0mM) against prostate (DU 145) cancer cell line. All the compounds have shown moderate (15.0–70.0 mM) to good (7.0–15.0mM) anticancer activity on both breast and neuronal cell lines [10] (Figure 3).

Figure 3 (a-f). All the compounds have shown moderate (15.0–70.0 mM) to good (7.0–15.0 mM) anticancer activity on both breast and neuronal cell lines.
M. Arshad et al. developed a series of tetrazolo-hydrazones and screened them against the ER+ve and ER-ve breast cancer cell lines which were respectively MCF-7, MDA-MB-231 and ZR-75. Out of the 14 compounds they synthesized,five compounds4a, 4b, 4c and 4dwere found to retard the growth of breast cancer cells. Based on the gene study the compounds 4b, 4c and 4dwere found more effective in retarding the growth of MCF-7 cells. While compound 4ashowed more growth retarding effects in ER negative MDA-MB-231 and ZR-75 cell lines. End-point determination was made by
MTT- colometric assay and the result for each compound wasreported as percent growth (GP %) of treated cells compared to uncontrolled growth [11] (Figure 4).

Figure 4. Compounds 4a, 4b, 4c and 4d were found to retard the growth of breast cancer cells.
Malani et al. in their research work synthesized some hybrid analogues by combining with tetrazole moieties. Almost, 20 compounds were synthesized and evaluated for anticancer activity against HepG2 and MCF-7 lines. Out of these compounds, three compounds5a, 5b and 5cexhibited very strong and a selective cytotoxic activity in the range of (IC50: 20.35-26.21 μM) against MCF-7 and (IC50: 10.14-20.32 μM) against HEPG-2. 5-Fluorouracil was taken as the positive control whose anti-proliferative activity against MCF-7 and HEPG-2 were respectively 10.48 μM and 05.36 μM [12] (Figure 5).

Figure 5. Compounds 5a, 5b and 5c exhibited very strong and a selective cytotoxic activity in the range of (IC50: 20.35-26.2 1 μM) against MCF-7 and (IC50: 10.14-20.32 μM) against HEPG-2. 5-Fluorouracil was taken as the positive control.
Kanakaraju et al. synthesized new series of 2, 5-disubstituted tetrazoles and 1, 2, 3-selena/thiadiazolyl-2H-tetrazole derivatives and evaluated for their in vitro cytotoxicity activities against Hep G2 and MCF-7 cell lines. Two of the compounds respectively 6awas found to be active against Hep G2 cell line (IC50: 48.83 L) and 6b exhibited good activity against both Hep G2 (IC50: 43.19 L) and MCF-7 (IC50: 47.15 L) cell lines [13] (Figure 6).

Figure 6. Two of the compounds respectively 6a was found to be active against Hep G2 cell line (IC50: 48.83 L) and 6b exhibited good activity against both Hep G2 (IC50: 43.19 L) and MCF-7 (IC50:47.15 L) cell lines.
E. Lukowska Chojnacka, et al. synthesized some series of new benzimidazole and benzotriazole derivatives containing a tetrazole moiety and cytotoxicity against human T-cell lymphoblast (CCRF-CEM) and breast adenocarcinoma (MCF-7) cell lines were evaluated using MTT assay. Two compounds mainly benzimidazole- terazole derivatives 7a and 7b exhibited highest toxicity at 25M concentration with complete (7a) or almost complete (7b) loss of cell viability after 48 h treatment in Leukemia cell line (CCRF-CEM). In case of Human breast adenocarcinoma cell line(MCF-7), compound 7aand 7b exhibited lower cytotoxicity than CCRF-CEM cell line but with higher compound concentration and prolongation of treatment time to 48 hours significantly decrease of cell viability was observed.Hence the introduction of aryl-tetrazole moiety into the alkyl side chain of the benzimidazole or benzotriazole resulted in higher cytotoxicity as compared to the nonsubstituted Tetrabromobenzimidazole (TBBi) and Tetrabromobenztriazole (TBBt) [14] (Figure 7).

Figure 7. Compounds mainly benzimidazole- terazole derivatives 7a and 7b exhibited highest toxicity at 25M concentration with complete (7a) or almost complete (7b) loss of cell viability after 48 h treatment in Leukemia cell line (CCRF-CEM).
Abd El Monaem et al. synthesized some new tetrazole-chalcone and tetrazole- pyrazoline hybrids which was further evaluated for in-vitro antiproliferative activity against cancer cell lines Vero-Bnormal cell line, colon HCT- 116, prostate PC-3cell lines and breast MCF-7cell lines using MTT assay.The activity appeared mostly with chalcone-tetrazole hybrids where Compound 8was found to be the most active antiproliferative agent against colon HCT-116 and prostate PC-3 cell lines (IC50 =0.6 and 1.6 μg/ml) with high selectivity indices in comparison with cisplatin (IC50 = 20 and 5 μg/ml) and 5-FU (IC50 = 17.3 and 21.4 μg/ml), respectively [15] (Figure 8).

Figure 8. Compound 8 was found to be the most active antiproliferative agent against colon HCT-116 and prostate PC-3 cell lines (IC50 =0.6 and 1.6 μg/ml) with high selectivity indices in comparison with cisplatin (IC50 = 20 and 5 μg/ ml) and 5-FU (IC50= 17.3 and 21.4 μg/ml).
Hou et al. synthesized some isatin-tetrazole hybrids and evaluated the compounds against breast (BT549 and MDA MB231), prostate (PC3 and DU145), and ovarian (PA1) cancer cell lines, by MTT assay. In his study he found that these hybrids could effectively inhibit the colony formation of DU145 cells and arrested the cells in the G2/M phase of the cell cycle in a dose dependent manner. Moreover, they can collapse the mitochondrial membrane potential and elevate intracellular ROS levels in DU145 cells.
Among them, two hybrids 9aand 9b (IC50: 21.49–96.14 μM) possessed a broad spectrum of activity against all tested cancer cell lines [7] (Figure 9). Zhang et al. in his review article describes about various tetrazole hybrids but out of all the hybrids he found three hybrids which have good anticancer activity against specific cell lines with IC50 values in nanomolar level, demonstrating the potential of tetrazole hybrids as anticancer candidates. In his review he mentioned that Agarwal, et al. examined some tetrazolesteroids hybrids as inhibitors of 5-reductase which is important in the treatment of benign prostate hyperplasia and found that hybrid 21c (IC50: 547 and 15.6 nM) emerged as a potent dual inhibitor for 5-reductase type 1 and 5-reductase type 2 isoezymes, and it possessed comparable activity to Finasteride (IC50: 453 and 40 nM) in vitro. Further, in-vivo 5-reductaseinhibitory studies showed significant reduction in rat prostate tumor weight (Figure 10 a).

Figure 9. Two hybrids 9a and 9b (IC50: 21.49–96.14 μM) possessed a broad spectrum of activity against all tested cancer cell lines.

Figure 10a. Comparable activity to Finasteride (IC50: 453 and 40 nM) in vitro.
Tetrazole-resveratrol (GI50: <0.01-5.28 μM) was a potent, selective, versatile and drug-like cytotoxic agent against both hematological and solid tumor cells (a panel of 60 human cancer cell lines including Leukemia, lung cancer, colon cancer, CNS, melanoma, ovarian cancer, renal cancer, prostate cancer and breast cancer cells), and GI50 values were <10 nM against over 90% of the tested cancer cell lines (Figure 10b).

Figure 10b. Tetrazole-resveratrol (GI50: <0.01-5.28 μM) was a potent, selective, versatile and drug-like cytotoxic agent.
Derivatives of pipedemic acid tetrazoles were also tested for in-vitro anti proliferative activity against cervix (SiHa), breast (MDA-MB-231) and pancreatic carcinoma cell lines by the sulforhodamine B (SRB) assay which was found more potent than the corresponding ciprofloxacin analogs [16] (Figure 10c).

Figure 10c. Derivatives of pipedemic acid tetrazoles were also tested for invitro.
In recent times, platinum based complexes like cisplatin, oxaliplatin, carboplatin have huge impact in the treatment of different cancers but due to their serious side-effects like neurotoxicity, ototoxicity, nephrotoxicity chemists now are trying to replace platinum with some other heavy metals like ruthenium, cobalt, gold, iron and cadmium along with the introduction of various heterocyclic ligands.
Abyar et al. in his report synthesized some cadmium complexes using 5- aminotetrazole and evaluated their cytotoxicity against MCF-7 (breast), Caco-2 (colorectal) and cisplatin-resistant A549 (lung) cancer cell lines. Tetrazole was selected as the ligand to form complex with cadmium because of its ability to decrease the toxicity of metals. In this study, cisplatin was used as the reference drug. The nephrotoxic potential of the compounds was evaluated in conditionally immortalized human proximal tubule epithelial cells (ciPTEC) which displayed a better cytotoxicity than cisplatin in the cancer cell lines with lower nephrotoxicity in ciPTEC (p<0.01) [17].
This new Cd-tetrazole complex like cisplatin, reduced the number of cells in G2/M phase of MCF-7 and A549 cells thereby acting as cell-cycle specific chemotherapeutic agents in vitro (Figure 11).
Tetrazole in anti-malarial activity
M. Tukulula et al. in his work synthesized some deoxyamodiaquine-tetrazole derivatives by incorporation of tetrazole moiety and protonable nitrogen into deoxyamodiaquine scaffold. Synthesized target compounds were evaluated forin- vitro anti-plasmodial activity against chloroquine-sensitive 3D7 strain and chloroquine- resistant K1 and W2-strains of Plasmodium falciparum. In-vitro primary screening of the compounds were tested against 3D7 strains at 6different concentrations (30, 10, 3, 1, 0.3, and 0.1 μg/mL). Compounds which affected the parasite growth at <lμg/mL were classified as active and further evaluated by 3-fold serial dilutions in a repeat test. The secondary screening were tested against both 3D-7 strain and K1 strain at 12 different concentrations with an appropriate starting concentrations based on thePrimary screen. The IC50 values for each parasite line were determined against CQ and other standard compounds appropriate for the assay. Compounds 12a, 12b and 12Cc (IC50 values ranging from 0.006 to 0.077μM) showed 2to 6-fold improvement in activity over amodiaquine and chloroquine against chloroquine-resistant K1 and W2 strains of Plasmodium falciparum [18] (Figure 12a-12c).

Figure 12. Compounds 12a, 12b and 12c (IC50 values ranging from 0.006 to 0.077μM) showed 2to 6-fold improvement in activity over amodiaquine and chloroquine against chloroquine-resistant K1 and W2 strains of Plasmodium falciparum.
Pandey et al. designed a series of novel 4-aminoquinoline-tetrazole derivatives by utilizing highly efficient TMSN3-Ugi multicomponent reaction and screened for their in-vitro antimalarial activities against both chloroquine-sensitive (3D7) and chloroquine-resistant (K1) strains of Plasmodium falciparum using a standardized inexpensive assay based on Malaria SYBR Green I nucleic acid staining dye based fluorescence (MSF) assay. Majority of these synthesized molecules exhibited promising in vitro antimalarial activity especially against CQ-R strain (K1). Compounds with significantin vitro activity were selected for in vivo activity in Swiss mice against with CQR N-67 strain of P. yoelli. Initially the in vivo activity of selected molecules was determined through intra-peritoneal route at the dose of 50 mg/ kg administered once daily for four consecutive days and monitored for parasitemia, and survival of mice until day 28 post infection. Out of all, two compounds mainly 13a and 13b showed in vivo suppression of 99.99% parasitaemia on day 4. Thus, Compound 13a and 13bshowed promising in vitro activity against both CQ-S as well as CQ-R strain of P. falciparum and also excellent in vivo antimalarial activity against P.yoelli which makes it potential for further development as potent antimalarial drug [19] (Figure 13 a and 13 b).

Figure 13. Compound 13a and 13b showed promising in vitro activity against both CQ-S as well as CQ-R strain of P. falciparum and also excellent in vivo antimalarial activity against P.yoelli which makes it potential for further development as potent antimalarial drug.
Lobo et al. in his work reported the synthesis of trioxolane–tetrazole conjugates and tetraoxane-tetrazole conjugates which were evaluated for both in-vitro and in-vivo antimalarial activity. In-vivo anti-malarial evaluation activity was carried out using the Peters’ 4-day suppressive test. Swiss albino mice were infected with Plasmodium bergheiNK65. In-vitro antimalarial activity were screened against chloroquine-susceptible strain (3D7) and multidrug-resistant (Dd2) P. falciparum strains using whole-cell SYBR Green I based assay.Animals were treated with 50 mg/ kg/day of each of the tested compounds (dissolved in PBS containing 1% DMSO), administrated orally, for four consecutive days. The control group was treated with a solution of PBS/1% DMSO. On days 5 (D5), 7 (D7) and 10 (D10) after parasite inoculation, tail blood smears were Giemsa-stained and examined microscopically to estimate parasitaemia.
Trioxolane–tetrazole conjugates 14aand 14b emerged as potential antimalarial candidates; showed nanomolar activity against ART-resistant P. falciparum parasites, negligible toxicity towards mammalian cells, totally suppressed P.berghei parasitaemia in mice and showed no cross resistance with CQ and ARTs [20] (Figure 14a and 14 b).

Figure 14.Trioxolane–tetrazole conjugates 14aand 14b emerged as potential anti-malarial candidates; showed nanomolar activity against ART-resistant P. falciparum parasites, negligible toxicity towards mammalian cells, totally suppressed P.berghei parasitaemia in mice and showed no cross resistance with CQ and ARTs.
Gao et al. in his work reported that incorporation of tetrazole into quinolone antimalarials showed more potent anti-plasmodial activity against drugresistant malaria.He carried his work with different hybrids such as tetrazolechloroquine hybrids, tetrazole-menadione hybrids, tetrazole-peroxide hybrids and in vitro anti- plasmodial activities of these hybrids were evaluated against chloroquine-sensitive 3D7,chloroquine-resistant K1 strains of P. falciparum, chloroquine-resistant W2 P. falciparum.
parasite inoculation, tail blood smears were Giemsa-stained and examined microscopically to estimate parasitaemia.
Trioxolane–tetrazole conjugates 14aand 14b emerged as potential antimalarial candidates; showed nanomolar activity against ART-resistant P. falciparum parasites, negligible toxicity towards mammalian cells, totally suppressed P.berghei parasitaemia in mice and showed no cross resistance with CQ and ARTs [20] (Figure 14a and 14 b).
Gao et al. in his work reported that incorporation of tetrazole into quinolone antimalarials showed more potent anti-plasmodial activity against drugresistant malaria.He carried his work with different hybrids such as tetrazolechloroquine hybrids, tetrazole-menadione hybrids, tetrazole-peroxide hybrids and in vitro anti- plasmodial activities of these hybrids were evaluated against chloroquine-sensitive 3D7,chloroquine-resistant K1 strains of P. falciparum, chloroquine-resistant W2 P. falciparum.
Majority of tetrazole-chloroquine hybrids compound 15exhibited excellent in vitro anti-plasmodial activities against chloroquine-sensitive 3D7 and chloroquine-resistant K1, W2 strains of P. falciparum with IC50 values in nanomolarlevel. The SAR revealed that position of tetrazole at phenyl, the type of linkers, substitutions on aromatic
Ring as well as substitutions on the tetrazole ring influenced the activity significantly (Figure 15 a and15 b).

Figure 15. Compound 15 exhibited excellent in vitro anti-plasmodial activities against chloroquine-sensitive 3D7 and chloroquine-resistant K1,W2 strains of P. falciparum with IC50 values in nanomolarlevel.
Three tetrazole-menadione hybrids 16 were evaluated for their potency against chloroquine resistant strain FcB1R of P. falciparum and glutathione reductase from P. falciparum. All hybrids (IC50: 0.9-1.4 μM) showed considerable activities against chloroquine resistant strain FcB1R of P. falciparum [21] (Figure 16).

Figure 16. Three tetrazole-menadione hybrids 16 were evaluated for their potency against chloroquine resistant strain FcB1R of P. falciparum and glutathione reductase from P. falciparum.
Tetrazole in anti-diabetic activity Momose et al. prepared a series of 5-(4-alkoxyphenyl- alkyl)-1H-tetrazole derivatives and evaluated their antidiabetic effects in two genetically obese and diabetic animal models, KKAy mice and Wistar fatty rats. One compound showed potent glucose lowering activity (ED25 = 0.0839 mg.kg−1·d−1) which was found to be 72 times more active than pioglitazone hydrochloride (ED25 = 6.0 mg·kg−1·d−1) in Wistar fatty rats [22].
Gao et al. synthesized some novel tetrazole-bearing N-glycosides as SGLT2 inhibitors which were further tested for in vivo hypoglycemic activity by mice oral glucose tolerance test (OGTT). The inhibition rates of blood glucose levels in mice OGTT for two compounds 17a and 17b were found to be 73.9% and 77.0% as compared with dapagliflozin (68.3%) which was taken as the positive control [23] (Figure 17).

Figure 17. Compounds 17a and 17b were found to be 73.9% and 77.0% as compared with dapagliflozin (68.3%) which was taken as the positive control.
Pegklidou et al. (2010) synthesized a novel series of pyrrole based on chemotypes and evaluated their activity as selective aldose reductase inhibitors. The two compounds 18a and 18bwere found as promising lead for the development of selective aldose reductase inhibitors, targeting the longterm complications of diabetes mellitus [24] (Figure 18 a and 18 b).

Figure 18. Compounds 18a and 18b were found as promising lead for the development of selective aldose reductase inhibitors, targeting the long-term complications of diabetes mellitus.
G. Navarrete-Vázquez et al. synthesized some tetrazoleisosteric analogue of clofibric acid by using a short synthetic route. The fibrates such as bezafibrate, clofibrate and fenofibrateare used as therapeutic agents in the treatment of diabetes complications in human which exerts a potent inhibitory activity against the enzyme 11-hydroxysteroid dehydrogenase type 1 (11-HSD1). Several lines of evidence have implicated 11-HSD1 activity in the etiology and/or maintenance of type 2 diabetes mellitus and metabolic syndrome. Glucocorticoids such as cortisol are potent antagonists to insulin action, promoters of gluconeogenesis in liver and 11-HSD1 catalyzes the conversion of inactive cortisone into the active hormone cortisol. Therefore, 11-HSD1 inhibitors are of considerable interest as potential treatments for type 2 diabetes. But the majority of fibrates are prodrugs, which are extensively metabolized by hydrolysis, leading to the free carboxylic acid form. So, in his work tetrazole was used as carboxylic acid bioisosteres which improves the bioavailability, chemical stability and increases the potency. The in-vitro assay of the compound 2-(4- chlorophenoxy)-2-methyl-N-(1Htetrazol- 5-yl) propanamide was determined by using a human embryonic kidney (HEK293) cell-based assay which showed 51.17%, of inhibition at 10 M. Compound 19 was also evaluated for in vivo hypoglycemic activity using a STZ–nicotinamide rat model of noninsulin dependent diabetes mellitus rat model. Glibenclamide (5 mg/Kg, p.o.) was taken as positive control. The hypoglycemic activity of compound 19 was determined using a 50 mg/Kg intragastric single dose. Compound 19 demonstrated important hypoglycemic activity, bylowering glycemia ranging from 16% to 77%. The effect was sustained during the 7 h of experiment and it was more pronounced than the hypoglycemic action showed by glibenclamide. It is important to mention that clofibric acid did not show any hypoglycemic effect [25] (Figure 19).

Figure 19. Compound 19 was also evaluated for in vivo hypoglycemic activity using a STZ–nicotinamide rat model of noninsulin dependent diabetes mellitus rat model. Glibenclamide (5 mg/Kg, p.o.) was taken as positive control.
Kaushik et al. in his work designed some tetrazole derivatives which was screened for in-vivo anti-diabetic activity by streptozocin induced-model in albino rat at a single dose of 200 mg/kg b.w per oral respectively for 7 days continuously. Blood was withdrawn from the tail vein each time. Blood glucose was measured at 0, 7th, 14th, 21stand 28th day by Blood Glucose. Out of all compounds one compound 20 showed good anti-diabetic activity with blood glucose lowering activity of 51.84% which was nearly with Rosiglitazone taken as the control [26] (Figure 20).

Figure 20. Compound 20 showed good anti-diabetic activity with blood glucose lowering activity of 51.84% which was nearly with Rosiglitazone taken as the control.
Tetrazole in antitubercular activity
Mycobacterium tuberculosis (MTB) is globally spread which is the main causative agent of tuberculosis. TB is the leading cause of death from a single infectious agent. Approximately 1.7 billion people have been infected with Mycobacterium tuberculosis globally whereas 5-10% of individuals develop TB disease during their lifetime.World Health Organization (WHO) estimated that 1.6 million people died from this disease in 2017, 8.6 million new cases and 1.3 million deaths occurred from TB infection around the world in 2012 [27]. Tuberculosis (TB) is pandemic and it is an infectious disease which claims hundreds of thousands of lives every year. Now days, although a reduction in new TB cases takes place, still the global dissemination of TB remains tremendous. Although a large number of antituberculosis drugs exist, the increasing occurrence of multidrug-resistant (MDR), extensively drug-resistant (XDR) and even totally drug resistant (TDR) TB strains highlights the necessity of a continual search for some novel highly efficient antimycobacterial compounds. To find out the importance of such investigations it is further emphasized by the relative toxicity and side effects of the drugs currently used for treatment, the low tolerability of the medicaments and the potential drug interactions of antituberculotics and antiretroviral drugs used in the treatment of HIV-positive TB patients. Several series of potential antituberculotics have been prepared and studied over the last few decades, and the number of these studies is still increasing [28].
P.B. M et al. Synthesized different tetrazole derivatives in a series having different aryl substituents on azatidinone core. The activity of all compounds againstM. tuberculosis strain H37Rv was assessed.Their activity mainly depends upon phenyl ring substituents.The Compounds with 4-methoxyphenyl and 4- dimethylamino phenyl unit, are highly active when compared with isoniazid and rifampin. In case of methoxy and dimehtylamino, the IC90 varies from 0.18 (21a) to 0.14 mM (21b).These novel molecules can be the promising antitubercular hit compounds [29] (Figure 21a and 21b).

Figure 21. Compounds with 4-methoxyphenyl and 4- dimethylamino phenyl unit, are highly active when compared with isoniazid and rifampin. In case of methoxy and dimehtylamino, the IC90 varies from 0.18 (21a) to 0.14 mM (21b). These novel molecules can be the promising antitubercular hit compounds.
George et al. synthesized some derivatives of 5-chloro-2-(5-(substituted phenyl)- 1H-tetrazol-1-yl) pyridines from the reaction of 2- amino pyridine derivative with substituted aromatic acid chlorides and sodium azide. The synthesized derivatives were evaluated for antitubercular activity by Microplate Alamar Blue assay (MABA) method.After the evaluation of the activity; it was found that the Compound (22) exhibited potential activity against Mycobacterium tuberculosis H37Rv [30] (Figure 22).

Figure 22. Compound (22) exhibited potential activity against Mycobacterium tuberculosis H37Rv.
Karabanovich G. et al. synthesized a series of novel 5-alkylsulfanyl-1-(3,5- dinitrophenyl)-1Htetrazoles. The derivatives with hydroxy, acetoxy, amino and acetamido groups instead of one of the nitro groups as well as derivatives with one trifluoromethyl group were prepared. The compounds were evaluated for the in vitro antimycobacterial activity which revealed very good activities for the 5-alkylsulfanyl- 1-(3,5-dinitrophenyl)-1H-tetrazoles against the drug-susceptible M.tb. My 331/88 strain, seven MDR/XDR strains of M.tb. and against the non-tuberculous strains M. kansasii My 235/80 and clinically isolated M. kansasii 6509/96. The direct attachment of the 3,5-dinitrophenyl moiety to the tetrazole nitrogen in the compounds of thatseries led to 2 - 4 times higher antimycobacterial efficiencies against M.tb. and strains of M. kansasii as compared to the parent compounds 1-alkyl/aryl-5- [(3,5- dinitrobenzyl)sulfanyl]-1H-tetrazoles. The MIC values were in the range of 0.25 - 1 μM, of the compounds of that series against M.tb [31] (Figure 23).

Figure 23. The 5-alkylsulfanyl- 1-(3, 5-dinitrophenyl)-1H-tetrazoles against the drug-susceptible M.tb. My 331/88 strain, seven MDR/XDR strains of M.tb. and against the non-tuberculous strains M. kansasii My 235/80 and clinically isolated M. kansasii 6509/96.
Roh et al. synthesized a series of tertiary amine which contain 3,5- dinitrophenyltetrazole derivatives. The synthesized derivatives were evaluated for in vitro antimycobacterial activity against CNCTC My 331/88 (H37Rv) and compared to the first-line drugs isoniazid (INH) and rifampicin (RIF). The MIC values of 1,4-dimethylpiperazine substituted, 4-methylmorpholine substituted, and 1- benzyl-4- methylpiperazine substituted compounds against M.tb. CNCTC My 331/88 (H37Rv) was found to be comparable to INH and RIF, ranging from 0.125 to 2 mM [32] (Figure 24).

Figure 24. MIC values of 1, 4- dimethylpiperazine substituted, 4-methylmorpholine substituted, and 1- benzyl-4- methylpiperazine substituted compounds against M.tb. CNCTC My 331/88 (H37Rv) was found to be comparable to INH and RIF, ranging from 0.125 to 2 mM.
Gao, et al. Reported that seven tetrazole-nitroimidazole hybrids were screened for their in vitro anti-TB activities against MTB H37Rv. All hybrids with minimum inhibitory concentration (MIC) values in a range of 0.313-5 μM were more potentthan kanamycin (MIC99: 5.40 μM), but no superior to streptomycin (MIC99: 0.27 μM). According to SAR the hybrids 25a-25c (MIC99: 0.313-0.625 μM) were generally more active than hybrids 25d-25g (MIC99: 0.313-5 μM). Compounds 25d- 25g contain quinoline fragment, was detrimental to the activity. With increasing length of the linker between tetrazole and quinoline resulted in reduced activity (Figure 25).

Figure 25. Compounds 25d- 25g contain quinoline fragment, was detrimental to the activity. With increasing length of the linker between tetrazole and quinoline resulted in reduced activity.
Among all the 7 hybrids the tetrazole-nitroimidazole hybrids 26 exhibited excellent in vitro anti-TB activity with MIC values ranging from 0.035-1.2, and conjugates 26a- 26c (MIC:0.035-0.29 μM) were more potent than the unsubstituted analog 25a (MIC:1.2 μM), which suggests that substituents at phenyl ring play a vital role in the activity. The most active hybrid 26d (MIC: 0.035 μM) was 14-fold more potent than Pretomanid (MIC: 0.50 μM) in vitro. Hybrid 26d also showed significantly greater in vivo efficacy than Pretomanid (4-fold) in a MTB acute infection mouse model, worth to be further investigated. Rest compounds can show moderate to weak antitubercular activity [33] (Figure 26).

Figure 26. Hybrid 26d also showed significantly greater in vivo efficacy than Pretomanid (4-fold) in a MTB acute infection mouse model, worth to be further investigated. Rest compounds can show moderate to weak antitubercular activity.
Tetrazole in antifungal activity
Fungal infections constitute a worldwide health problem and the increase in the number of human diseases caused by pathogenic fungi. These infections are difficult to treat and especially dangerous for elderly people and patients with an impaired immune system due to neutropenia, AIDS, diabetes, cancer and induced immunosuppression after an organ transplant. Among many types of infections the most common are these caused by Candida spp., Aspergillus spp. and Cryptococcus spp., which lead to candidiasis, aspergillosis and cryptococcosis, respectively. The most frequent fungal pathogens are Candida albicans, Aspergillus fumigatus, Aspergillus niger and Cryptococcus neoformans. The currently used antifungal drugs display various mechanisms of action. Azoles (e.g., Ketoconazole, Itraconazole, Fluconazole, Voriconazole, Posoconazole, Ravuconazole), and allylamines (e.g. Terbinafine) inhibit synthesis of ergosterol, which is the major component of the fungal cell membrane. Polyene antibiotics (e.g., Amphotericin B, Nystatin) interact with ergosterol and thus disrupt the fungal membrane and increase its permeability.Other compounds inhibit synthesis of the major components of the cell wall e.g. inhibitors of glucan synthesis (e.g., Echinocandins, Caspofungin, Micafungin) and inhibitors of chitin synthesis (e.g., Nikkomycin, Polyoxins).We also know antifungal agents that inhibit synthesis of nucleic acids (e.g. Flucytosine) or proteins (e.g. Sordarins) and many others with a more complex mechanism of action. The treatment of fungal infections is problematic because of an emerging drugs resistance in fungi; therefore studies on the design and synthesis of new effective antifungal compounds are conducted and focus on tetrazole derivatives. These azoles significantly inhibit the growth of many pathogenic strains [34].
Upadhayaya R.S. et al. Synthesized some novel tetrazole based triazole derivatives. The antifungal activity of these new compounds was evaluated by in vitro agar diffusion and micro broth dilution assay. Most of the compounds showed activity against fungal cultures at 500 μg ml–1 concentrations of compounds in the agar diffusion assay. These compounds were evaluated by micro broth dilution assay to determine the minimum inhibitory concentration (MIC) values. The MIC values were calculated(in μg ml–1) against Candida species, C. neoformans and Aspergillus species in comparison with fluconazole and itraconazole. Compound 27a having 2-butoxy substitution on the phenyl ring of piperazine was the most active compound with MIC value of 1.0–2.0 μg ml–1 for Candida albicans, Candida tropicalis, Candida parapsilosis and C. neoformans. Improvement in the spectrum and antifungal activity was observed in compounds having methyl group at C-3 position [35] (Figure 27).

Figure 27. Compound 27a having 2-butoxy substitution on the phenyl ring of piperazine was the most active compound with MIC value of 1.0–2.0 μg ml–1 for Candida albicans, Candida tropicalis, Candida parapsilosis and C. neoformans. Improvement in the spectrum and antifungal activity was observed in compounds having methyl group at C-3 position.
Dhayanithi V, et al. synthesized a novel series of 5-thio substituted tetrazole derivatives.All the synthesized compounds were tested for its antifungal activity.The study was carried out against Aspergillus flavus, Aspergillus fumigatus, Penicilliummarneffei and Trichophytonmentagrophytes and compared with the standard itraconazole drug. The zone of inhibition in mm and MIC in mg mL–1 were determined for the synthesized compounds. Most of the compounds exhibited good antifungal activity, but among all the tested compounds compounds 28 and 29 showed the highest antifungal activity against all four fungal strains, namely A. flavus, A. fumigatus, P. marneffei and T. menta-grophytes [36] (Figures 28 and 29).

Figure 28. Compounds 28 showed the highest antifungal activity against all four fungal strains, namely A. flavus, A. fumigatus, P. marneffei and T. mentagrophytes.

Figure 29. Compounds 29 showed the highest antifungal activity against all four fungal strains, namely A. flavus, A. fumigatus, P. marneffei and T. mentagrophytes.
Libero FM, et al. Synthesized novel selenium and tellurium-containing tetrazoles which is a class of chalcogen compounds. From all the synthesized compounds compound 30 was screened for antifungal activity and presented inhibitory property against seven fungal strains like C. albicans, C. lipolytica, C. Parapsilosis, C. Laurentii, T. Asahii, C. Guilhermondi, C. globosa. It was observed that the compound 4a showed good results on inhibition of T. asahii and C. lipolytica [37] (Figure 30).

Figure 30. Compound 30 was screened for antifungal activity and presented inhibitory property against seven fungal strains like C. albicans, C. lipolytica , C. Parapsilosis, C. Laurentii, T. Asahii, C. Guilhermondi, C. globosa.
Malik M.A. et al. synthesized some novel tetrazole ring containing acyl hydrazone. The acyl-hydrazone derivatives (31a–31j) were obtained through a condensation reaction of 2-[5-(4-chlorophenyl)-1H-tetrazol-1-yl]- acetohydrazide with different aromatic aldehydes in ethanol medium in 1:1 molar ratio. The antifungal activity was evaluated for all the synthesized compounds. MIC (Minimum Inhibitory Concentration) and MFC (Minimum Fungicidal Concentration) were determined of 3 standard laboratory Candida isolates, 11 clinically obtained Candida isolates and 3 resistant strains against all the test compounds. It is found that compounds 31c–31g showed fungicidal activity while compounds 31a, 31h-31j showed fungistatic activity as that of the most commonly used antifungal fluconazole. Compound 31b is inactive against all the tested isolates of Candida [38] (Figure 31).

Figure 31. The acyl-hydrazone derivatives (31a–31j) were obtained through a condensation reaction of 2-[5-(4-chlorophenyl)-1H-tetrazol-1-yl]- acetohydrazide with different aromatic aldehydes in ethanol medium in 1:1 molar ratio.
Chojnacka EL, et al. synthesized new 2,5-disubstituted tetrazole derivatives containing benzothiazole, benzoxazole or phenylsulfonyl moiety were synthesized by N-alkylation of aryltetrazole with 2-[(3-chloropropyl) sulfanyl]- 1,3-benzothiazole or 2-[(3-chloropropyl)sulfanyl]-1,3-benzoxazole and Michael-type addition of aryltetrazole to phenyl vinyl sulfone. The compounds were tested against the moulds: Fusarium sambucinum, Fusarium oxysporum, Colletotrichum coccodes, Aspergillus niger, and the yeast Candida albicans. The synthesized compounds exhibit moderate to high antifungal activity against C. coccodes MC 1 and C. albicans ATCC 90028, respectively. It was also reported that the benzoxazole derivative 32c can modify the cell wall architecture. The benzoxazole derivatives 32c and 32d exhibited activity against C. albicans at concentrations that are nontoxic to the invertebrate host G. mellonella, which should give insight into the clinical relevance of this phenomenon [34] (Figure 32).

Figure 32. The benzoxazole derivative 32c can modify the cell wall architecture. The benzoxazole derivatives 32c and 32d exhibited activity against C. albicans at concentrations that are non-toxic to the invertebrate host G.
Tetrazole in anti-inflammatory activity
Inflammation constitutes a major health problem and for the treatment of this mainly nonsteroidal anti-inflammatory drug (NSAID) is used. But it is seen that approximately 1, 07,000 patients are hospitalized annually for nonsteroidal anti- inflammatory drug (NSAID)-related gastrointestinal (GI) complications and at least 16,500 NSAID-related deaths occur each year among arthritis patients alone. So due to the serious drawback of all analgesic and anti-inflammatory drugs that is acidity problems which limits their use in many cases. For this reason most of the patients were unable to continue these drugs for their ailment from diseases. To overcome the drawback of anti-inflammatory drugs several newer lead molecules were selected which include indole nucleus, arylalkyl acid nucleus, pyrazolone, indan etc. and attempt has been taken to discover novel anti-inflammatory agent without or less gastrointestinal effects [39]. Some structural or molecular modifications were done in these nuclei either by introducing functional groups or ring fusion. Both the modifications led to many promising anti inflammatory agents. It was already established that tetrazole, which is an aromatic azapyrrole group, is metabolically stable [40]. It has acidic characteristics closely similar to that of the carboxylic group. Ithas been also reported that anti inflammatory and related biological activities have been improved or abolished by the substitution of a 5-tetrazole group in place of carboxyl function [41].
Bepary S, et al. Synthesized a number of indanyltetrazole derivatives like 5- (6′-chloroindan-1′-yl)tetrazole (CIT), 5-(6′-bromoindan-1′-yl)tetrazole (BIT), 5-(6′- chloroindan-1′-yl)methyltetrazole (CIMT), 5-(6-romoindan-1′- yl)methyltetrazole(BIMT).All the synthesized compounds were evaluated for the anti inflammatory activity in carragennan induced rat paw edema in Swiss albino Wister rats for 24-hour period at the dose of 100 mg/kg of body weight by intraperitoneal route. The drug phenylbutazone (PBZ) was used as the standard. It was observed that all the compounds exhibited inhibition on rat paw edema with peak actions after 3 hours of administration. Compounds CIMT and BIMT were further evaluated at dose of 50 mg/kg of body weight where CIMT showed higher activity than others [42] (Figure 33).

Figure 33. Synthesized a number of indanyltetrazole derivatives like 5- (6′-chloroindan-1′-yl) tetrazole (CIT), 5-(6′-bromoindan-1′-yl) tetrazole (BIT), 5-(6′- chloroindan-1′-yl) methyltetrazole (CIMT), 5-(6′-bromoindan-1′- yl) methyltetrazole (BIMT).
Mohite PB, et al. Synthesized some novel N-substituted tetrazoles. Their work deals with the reaction of 5-phenyl tetrazole with acetic anhydride to yield 5-phenyl 1- acetyl tetrazole which on further reaction with different aromatic aldehydes forms chalcones. Chalcones on further reaction with isoncotinic acid hydrazide affords pyrazolines. The synthesized compounds are screened for anti-inflammatory activity by using inhibition of albumin denaturation technique. The ibuprofen was used as standard drug. The standard drug ibuprofen and test compounds were dissolved in minimum amount of dimethyl formamide (DMF) and diluted with phosphate buffer (0.2 M, pH 7.4). Final concentration of DMF in all solutions was less than 2.0%. Test solution (1 ml) containing 0.2 mM conc. of drugs was mixed with 1 ml of 1% mM albumin solution in phosphate buffer and incubated at 270 +10C in BOD incubator for 15 min. Denaturation was induced by keeping the reaction mixture at 600 + 10C water bath for 10 min. After cooling the turbidity was measured at 660 nm (UV- Visible Spectrophotometer Jasco V-630). Percentage of inhibition of denaturation was calculated from control where no drug was added. After the evaluation it was found that the standard drug Ibuprofen exhibited 90.00% inhibition of albumin denaturation. Among all the compounds, compounds 34a and 34b inhibit the denaturation of albumin in 68.33% and 70.00% respectively when compared with control possess potent anti-inflammatory activity [43] (Figure 34).

Figure 34. Compounds 34a and 34b inhibit the denaturation of albumin in 68.33% and 70.00% respectively when compared with control possess potent anti-inflammatory activity.
Kumbar M.N, et al. designed a series of novel 5-(1-aryl-3-(thiophen-2-yl)-1Hpyrazol- 4-yl)-1H-tetrazoles. To evaluate the anti-inflammatory property of synthetic drugs Murine macrophage cell line RAW264.7 has been employed. The synthetic compounds were tested against RAW mouse murine cancer cell line at three different concentration levels viz., 1.00, 5.00 and 10.00 mg/ml. The standard drug Aminoguanidine at 200 mM concentration can inhibit the production of Nitric oxide (NO) which is formed during activity. The synthetic compound inhibited the NO production at dose dependent manner. Among all the compounds, compounds 35a-f has significantly inhibited the nitric oxide production and also they can reduce the cell viability. Thus, it is concluded that the compounds can show potent anti- inflammatory property by inhibiting the NO production. The synthetic compounds reduced the Lipopolysaccharide (LPS)-induced NO production by both through reduction of iNOS protein levels and also through post-translational modifications [44] (Figure 35).

Figure 35. Synthetic compounds reduced the Lipopolysaccharide (LPS)- induced NO production by both through reduction of iNOS protein levels and also through post-translational modifications.
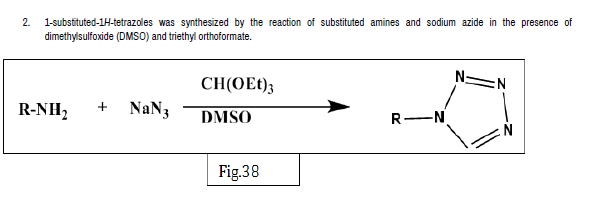
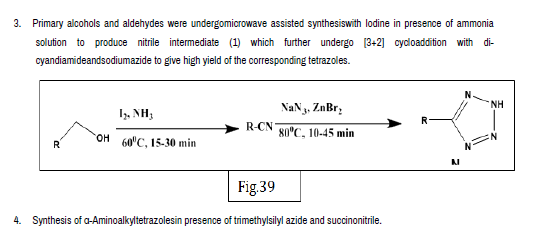
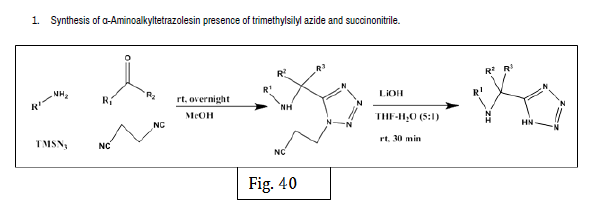
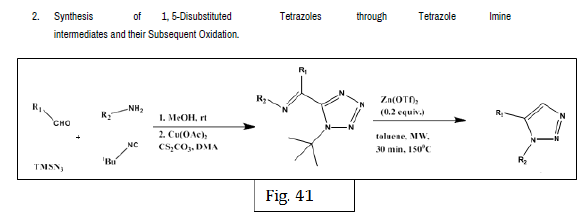
Rajasekaran A, et al. designed a series of novel 10-[(1-acyl-1-H-tetrazol-5- yl)]-10-H-phenothiazines and synthesized by reacting 10-[1-H-tetrazol-5-yl)]- 10-H- phenothiazines with desired acylating or sulfonating agents. The antiinflammatory activity was evaluated by carrageenan induced rat paw edema method. Test compounds were administered intraperitoneally in a dose of 25 mg/kg body weight. The anti-inflammatory activity was determined in terms of percentage inhibition of edema of each group against the control group. From the 12 synthesized compounds, compounds 36a and 36b showed potent anti-inflammatory activity [45] (Figure 36-41).
Synthetic Schemes ForTetrazole Derivatives
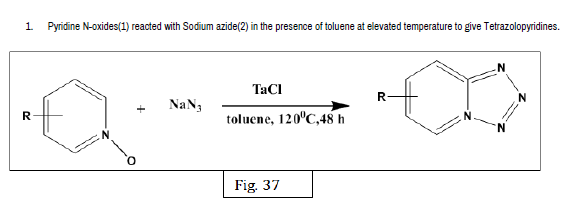
Malaria Control & Elimination received 1187 citations as per Google Scholar report