Mini Review - (2023) Volume 12, Issue 4
Received: 01-Aug-2023, Manuscript No. MBL-23-111524;
Editor assigned: 02-Aug-2023, Pre QC No. P-111524;
Reviewed: 14-Aug-2023, QC No. Q-111524;
Revised: 19-Aug-2023, Manuscript No. R-111524;
Published:
28-Aug-2023
, DOI: 10.37421/2168-9547.2023.12.390
Citation: Pobezinskaya, Elena. “Molecular Crystals is the Preparation of the Initial Structure of Molecules.” Mol Bio 12 (2023): 390.
Copyright: © 2023 Pobezinskaya E. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Ferroelectric materials are materials that exhibit a spontaneous electric polarization in the absence of an external electric field. These materials are of great interest for applications in electronic devices such as memory storage, data processing, and sensing. The study of ferroelectric materials has been a subject of intense research in recent years. Theoretical and computational methods have played a significant role in the understanding of the properties and behavior of these materials. Molecular dynamics simulations are a powerful tool for the study of ferroelectric materials. In this article, we will discuss the use of molecular dynamics simulations to study the ferroelectric properties of diisopropyl- ammonium halide molecular crystals.
Crystals • Molecules • Ferroelectric materials • Molecular dipoles • Behavior of atoms
Di-isopropyl-ammonium halide molecular crystals are a class of materials that exhibit ferroelectric properties. These materials have attracted attention due to their interesting properties such as high dielectric constant, large electro-optic coefficients, and excellent optical properties. Molecular dynamics simulations can be used to study the structural and dynamic properties of these materials at the atomic level. Molecular dynamics simulations are a computational method that allows the study of the motion and behavior of atoms and molecules over time. The simulations use classical mechanics to calculate the positions and velocities of atoms and molecules as they interact with each other. The interatomic interactions are described by a potential energy function, which includes terms that describe the bonding between atoms and the non-bonded interactions such as van der Waals and electrostatic interactions. The first step in the molecular dynamics simulation of di-isopropyl-ammonium halide molecular crystals is the preparation of the initial structure. The crystal structure of the material is obtained from experimental data or from theoretical calculations [1].
The structure is then optimized using a force field to obtain the equilibrium geometry. The force field is a mathematical function that describes the interatomic interactions in the material. In order to perform MD simulations of DIPA molecular crystals, we first need to develop an accurate model of the system. This typically involves developing a force field that describes the interactions between the atoms and molecules in the system. Once the initial structure is prepared, the simulation is started by specifying the initial positions and velocities of the atoms. The simulation is then run for a certain period of time, during which the positions and velocities of the atoms are updated based on the equations of motion. The simulation time can range from picoseconds to microseconds or even longer, depending on the system size and the computational resources available. The output of the simulation includes the trajectories of the atoms, which can be used to study the structural and dynamic properties of the material [2].
The trajectory data can be analyzed using various techniques such as radial distribution functions, mean-square displacements, and vibrational spectra. In the case of di-isopropyl-ammonium halide molecular crystals, molecular dynamics simulations have been used to study the ferroelectric properties of the material. The ferroelectric properties arise due to the ordering of the molecular dipoles in the crystal. The dipole moments of the molecules are aligned in a particular direction, resulting in a net polarization. The molecular dynamics simulations have been used to study the effect of temperature on the ferroelectric properties of di-isopropyl-ammonium halide molecular crystals. The simulations have shown that the polarization of the material decreases with increasing temperature. This is due to the thermal motion of the molecules, which disrupts the alignment of the dipole moments. The simulations have also been used to study the effect of external electric fields on the ferroelectric properties of the material. The simulations have shown that the polarization of the material can be reversed by applying an external electric field in the opposite direction [3].
This phenomenon is known as ferroelectric switching and is of great interest for applications in memory storage and data processing. The molecular dynamics simulations have also been used to study the structural properties of di-isopropylammonium halide molecular crystals. The simulations have shown that the crystal structure of the material is strongly influenced by the interactions between the molecules. Ferroelectric materials are of great interest in the field of electronics, as they can exhibit spontaneous polarization that can be switched on and off through the application of an external electric field. This property makes them ideal for use in memory storage devices, capacitors and sensors. Di-isopropylammonium halide molecular crystals are a class of ferroelectric materials that have been studied extensively for their unique properties. In this article, we will discuss the use of molecular dynamics simulations to study Ferro electricity in DIPA molecular crystals. Molecular dynamics is a computational technique that is widely used to study the behavior of atoms and molecules in a system [4].
In MD simulations, the positions and velocities of atoms are tracked over time using classical mechanics. This allows us to study the movement of atoms and molecules, as well as the interactions between them, in great detail. MD simulations can be used to study a wide range of systems, including solids, liquids and gases. These phases are characterized by different patterns of polarization within the crystal, and can be switched between by the application of an external field. In recent years, MD simulations have become an increasingly popular tool for studying ferroelectric materials. This is because MD simulations allow us to study the behavior of ferroelectric materials at the atomic level, which can provide valuable insights into the mechanisms that govern their properties. In particular, MD simulations can be used to study the structural and dynamical properties of ferroelectric materials, as well as the effect of external fields on their behavior [5].
DIPA molecular crystals are a particularly interesting class of ferroelectric materials, as they exhibit a range of unique properties. For example, DIPA crystals can exhibit a range of different ferroelectric phases, depending on the temperature and pressure of the system. In addition, DIPA crystals are known to exhibit a range of structural and dynamical properties that are related to their ferroelectric behavior. One of the key advantages of using MD simulations to study DIPA molecular crystals is that they allow us to study the behavior of these materials at the atomic level. This can provide valuable insights into the mechanisms that govern their ferroelectric behavior. For example, MD simulations can be used to study the effect of external fields on the polarization of DIPA crystals, which can provide insight into the mechanisms that govern the switching behavior of these materials. The force field is typically developed by fitting to experimental data, such as X-ray diffraction and spectroscopic measurements. Once the force field has been developed, we can perform MD simulations of the system using standard techniques [6].
One of the key challenges in performing MD simulations of DIPA molecular crystals is that these materials can exhibit a range of different phases depending on the temperature and pressure of the system. This means that we need to carefully control the conditions of the simulation in order to ensure that we are studying the correct phase of the material. In addition, we need to ensure that the simulation is long enough to capture the relevant dynamics of the system. Despite these challenges, MD simulations have been used successfully to study a range of properties of DIPA molecular crystals. For example, MD simulations have been used to study the structural properties of these materials, including the formation of domains and the behavior of defects. In addition, MD simulations have been used to study the dynamics of the system, including the behavior of the polarization and the effect of external fields on the system. One particularly interesting aspect of DIPA molecular crystals is their ability to exhibit a range of different ferroelectric phases.
None
None
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
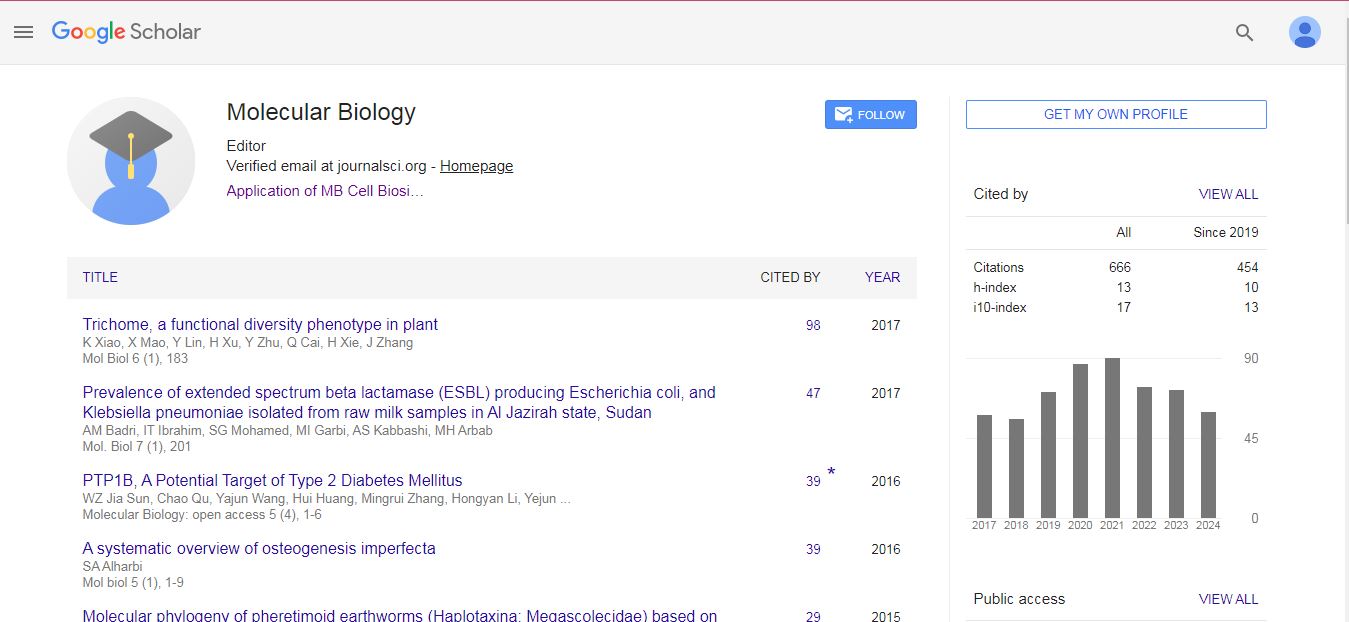
Molecular Biology: Open Access received 607 citations as per Google Scholar report