Mini Review - (2023) Volume 13, Issue 1
Received: 11-Jan-2023, Manuscript No. JBL-23-86694;
Editor assigned: 13-Jan-2023, Pre QC No. JBL-23-86694 (PQ);
Reviewed: 27-Jan-2023, QC No. JBL-23-86694;
Revised: 03-Feb-2023, Manuscript No. JBL-23-86694 (R);
Published:
10-Feb-2023
, DOI: 10.37421/2165-7831.2023.13.296
Citation: Masuda, Tatsuya Tomoo Daifu, Masamitsu Mikami and Hiroshi Sugiyama, et al. "Cluster Regulation of RUNX (CROX): Strategy against Malignant Rhabdoid Tumor". J Blood Lymph13 (2023) 296.
Copyright: © 2023 Masuda T, et al. This is an open-access article distributed under the terms of the creative commons attribution license which permits unrestricted
use, distribution and reproduction in any medium, provided the original author and source are credited.
Malignant Rhabdoid Tumor (MRT) is a quite rare malignant pediatric disease that occurs primarily in infants and young children. Although most patients with MRT are treated with the intensive multimodal treatment, the results are unsatisfied and there is no established standard of care for MRT. Therefore, there is a great need for more effective treatment(s) for MRT. Recently, we have demonstrated that RUNX1 silencing and “CROX (Cluster Regulation of RUNX)” strategy using our novel RUNX inhibitor (Chb-M') strongly reduce MRT cell proliferation rate via the augmentation of p53-mediated apoptotic pathway in vitro and in vivo. In this mini-review, we describe the molecular characteristics of our RUNX inhibitor, DNA-alkylating Pyrrole-Imidazole (PI) polyamides, and its anti-tumor effects on a variety of cancers including MRT. Finally, we discuss the precise molecular mechanisms behind p53-mediated apoptosis induced by RUNX inhibition in MRT cells.
Malignant rhabdoid tumor • RUNX1 • Regulation of gene expression • Polyamide • p53
Malignant Rhabdoid Tumor (MRT) is a quite rare malignant disease that occurs primarily in infancy [1]. MRT commonly arises in the central nervous system (ATRT, atypical teratoid/rhabdoid tumor), and also takes place in other parts of the body such as kidneys, liver, neck and thorax. Nearly all of MRT genetically carry loss of function mutations in SMARCB1 (hSNF5/INI1), a potent tumor suppressor gene [2]. SMARCB1 encodes a core subunit of SWI/SNF chromatin-remodeling complex. SWI/SNF complex takes advantage of the energy of ATP hydrolysis to remodel chromatin and thus modulates transcription of its target genes. It has been suggested that SMARCB1 is one of the major genes involved in the development of MRT. Wang et al. described that oncogenesis in the absence of SMARCB1 occurs regardless of SWI/SNF complex inactivation, but depends on the activity of SMARCA4 (BRG1), ATPase subunit of SWI/SNF complex [3].
Currently, most patients with MRT are subjected to the intensive multimodal treatments, combining with the early surgical resection of primary tumor, chemotherapy, radiotherapy or High Dose Chemotherapy (HDCT) followed by autologous stem-cell rescue [4]. However, the treatment result is not always enough, and there is no established standard therapy for MRT. Therefore, more effective treatment modality against MRT is highly desired.
Runt-related Transcription Factor 1 (RUNX1), also known as Acute Myeloid Leukemia 1 protein (AML1), is a member of the corebinding factor family of transcription factors and indispensable for the establishment of definitive hematopoiesis [5]. RUNX1 is one of the most frequently mutated genes in a variety of hematological malignancies. However, the current studies strongly indicate that wild-type RUNX1 is required for proliferation and survival of certain types of leukemia cells [6]. Moreover, not only RUNX1 but also the entire RUNX family (RUNX1, RUNX2 and RUNX3) serve as master regulators of the multistep progression to malignancy through the crosstalk with the key signaling pathways [7]. Thus, we have focused on the potential role of RUNX in cancer and found that RUNX is implicated in growth of various cancers including leukemia and gastric cancer, and that RUNX directly and/or indirectly regulates the proliferative signaling such as Receptor Tyrosine Kinase (RTK) and BCR-ABL1, and the tumor suppressor p53 [8-10]. Notably, it has been reported that SWI/SNF complex interacts with RUNX111. Unfortunately, it remains elusive how RUNX1 could participate in the development of MRT. We have recently described that RUNX1 silencing and our novel RUNX inhibitor (Chb-M’) [8,11]. Efficiently suppress the proliferation of MRT cells through the augmentation of p53-dependent apoptotic pathway in vitro and in vivo [12,13]. In this mini-review, we describe the anti-tumor effects of our RUNX inhibitor, and also the functional importance of p53-mediated proapoptotic pathway potentiated by RUNX inhibition in MRT cells.
DNA-alkylating PI polyamide targeting RUNX transcription factor genes
According to our previous studies, depletion of RUNX1 alone effectively suppressed AML cells, and moreover, the comprehensive knockdown of RUNX1, RUNX2 and RUNX3 led to a marked reduction of their proliferation rate compared to RUNX1 depletion alone [8]. Upon knockdown of RUNX1, the expression levels of RUNX2 and RUNX3 became increased. Similarly, the simultaneous depletion of RUNX2 and RUNX3 caused a significant increase in RUNX1 level. We referred this mechanism as to “genetic compensation of RUNX family”14. Among RUNX family members, we have knocked down RUNX1 alone in MRT [12,13]. Based on our compensation theory, it is of great interest to ask the possible roles of other members of RUNX family (RUNX2 and RUNX3) in MRT.
The presence of the compensation mechanism among RUNX family members prompted us to imagine that targeting whole RUNX family could be an effective strategy to suppress malignant phenotypes of leukemia cells. We referred this strategy as to “CROX (Cluster Regulation of RUNX)” [14,15]. To evaluate CROX strategy, we have developed a novel RUNX inhibitor termed Chb-M’ [8,15,16]. Chb-M’ is a Pyrrole-Imidazole (PI) polyamide interlocked with a hairpin conjugated with alkylating reagent chlorambucil that specifically targets consensus RUNX-binding sequence, 5’- TGTGGT-3’. Since Chb-M’ specifically prevents the binding of RUNX family members to their consensus binding sequence, Chb- M’ efficiently switches off RUNX-target genes. As expected, our previous study10 indicates that DNA alkylation mediated by chlorambucil might exert an important role during the transcriptional regulation under our experimental conditions.
From our previous studies, Chb-M' displayed a remarkable antitumor effect on numerous cancers such as AML, Ph+ ALL, lung cancer, HER2-positive gastric cancer and MRT in vitro and in vivo [8,10,12,13]. Of note, we have revealed that Chb-M' significantly suppresses the proliferation signals such as RTK and BCR-ABL1, and induces p53-mediated apoptosis, which is similar to the results obtained from silencing of RUNX genes. In Ph+ ALL, we have confirmed on-target for the promoter region of BCR-ABL1 gene by a ChIP-PCR experiment using alkylating agent-free Chb-M’ (Simple- M’)[10]. For MRT, Chb-M’ was highly effective on all three MRTderived cell lines examined (IC50 value at 72 h: MP-MRT-AN; 0.13 μM, KP-MRT-RY; 1.64 μM, KP-MRT-YM; 0.16 μM) [12]. In Cell Line- Derived Xenograft (CDX) or Patient-Derived Xenograft (PDX) models, Chb-M' (320 μg/kg body weight injections twice per week, the generally accepted dose of PI polyamides) attenuated tumor growth more than the existing drugs (AraC for AML, imatinib for Ph+ ALL, gefitinib for lung cancer and lapatinib for HER2-positive gastric cancer) or Chb-Scramble (Chb-S), which is a PI polyamide targeting 5’-WGGCCW-3’ sequence and randomly binds to DNA, suggesting the benefits of targeting RUNX-binding sequences [8]. Next, we have checked the pharmacological safety of Chb-M' (320 μg/kg body weight injections twice per week for 3 months) using wild-type C57BL/6 mice. Our results showed that no side effects are observed in the complete blood counts of peripheral blood and body weight [8]. Since RUNX genes are involved in hematopoiesis, we also examined the possible effects on immature stem cells in bone marrow. Based on our results, Chb-M' did not change the proportion of stem cells and did not affect their ability to proliferate and differentiate as examined by a colony-forming cell assay [8]. Furthermore, CDX experiments using gastric cancer cells demonstrated the cancer cell-specific uptake of Chb-M'8. The obvious accumulation and retention of alkylating agent-conjugated polyamides in cancer cells might contribute to their anti-tumor effects, and no aberrant effect on hematopoietic stem cells was detectable. In addition, Chb-M' reduced the engraftment of AML cells to the bone marrow vascular niche through the regulation of the adhesion-related E-selectin as well as the resultant anti-tumor effect [15]. Since RUNX is implicated in various signals other than proliferation signals [7]. We have a plan to investigate not only growth control but also anti-tumor immunity and metastasis control. For cancers in which CROX strategy does not work, we are willing to examine whether Chb-M' could synergize with common anticancer agents to enhance anti-tumor immunity or suppress metastasis.
Induction of p53-mediated apoptosis via RUNX depletion in MRT
We have found for the first time that RUNX1-BCL11A/TRIM24 regulatory axis is one of the molecular mechanisms behind p53- mediated apoptosis induced by RUNX1 depletion in AML [8]. BCL11A and TRIM24 were extracted by creating Venn diagrams using genes that correlate with RUNX1 and genes that display RUNX1 ChIP-seq peaks in AML public data. Both of them encode oncoproteins which suppress p53. BCL11A regulates the expression of Bcl2, Bcl2-xL and Mdm2, which inhibit p53 activity [17]. While, TRIM24 negatively regulates p53 level through the ubiquitination, suggestive that TRIM24 could be a potential therapeutic target to restore tumor-suppressive activity of p53 [18]. The actual binding of RUNX family to several RUNX-binding consensus sites (mainly 5′-TGTGGT-3′ and rarely 5′-TGCGGT-3) of their promoter regions was verified by ChIP-PCR experiments. Both RUNX1 silencing and Chb-M' treatment decreased their expression at both mRNA and protein levels. Consistent with these observations, we have demonstrated that forced expression of these genes rescues the proliferation rate of RUNX1-knockdown AML cells. Only TRIM24 has been confirmed in MRT [13].
As for the functional importance of p53 in MRT, Howard et al. described that MDM2 and MDM4, the putative negative regulators of p53, might be suitable therapeutic targets for MRT [19]. MDM2 and MDM4 were identified by the comprehensive CRISPR-Cas9 screening for MRT. When compared to other tumor cells with wildtype p53, inhibitors against MDM2/MDM4 were much more effective on MRT, indicative that loss of function in SMARCB1 contributes to their sensitivity. Recently, Carugo et al. found that MRT has no somatic mutations of p53, and the deficiency in SMARCB1 potentiates pro-apoptotic p53 pathway in MRT [20]. Their findings imply that the activation of p53-mediated pro-apoptotic pathway is a promising therapeutic target for MRT, and basically supported our recent results showing that depletion of RUNX induces p53- dependent apoptosis [12,13].
However, it is worth noting that the activation of p53 pathway alone is not sufficient to control tumor cell proliferation, because p53 has an ability to protect tumor cells from the proteotoxic cell death by promoting autophagy [20]. In this report, BIRC5, which belongs to Inhibitor of Apoptosis (IAP) family, has been shown to participate in an anti-apoptotic mechanism. According to their results, in SMARCB1-deficient murine and human tumors, IAP protein BIRC5 was dramatically upregulated and then elevated p53 threshold that causes apoptotic cell death. By using Human Apoptosis Array Kit, we have recently confirmed that both RUNX1 knockdown and Chb-M' treatment reduce BIRC5 [13]. Moreover, the rescue experiments revealed that forced expression of BIRC5 effectively rescue RUNX inhibition-dependent attenuation of cell proliferation, indicating that BIRC5 might also participate in the suppression mechanism.
Thus, it is likely that the decrease in BIRC5 might promote apoptotic cell death triggered by an increase in p53. Taken together, we have discovered an important role of RUNX transcription factor during p53-mediated apoptotic cell death and proliferation of MRT cells. “CROX (Cluster Regulation of RUNX)” could be a therapeutic strategy against MRT, and also our novel RUNX inhibitor, Chb-M’, could be a potential drug for patients with multimodal treatments-resistant refractory MRT through the potentiation of p53-mediated apoptotic pathway. In this review, we have described the efficacy of our “CROX (Cluster Regulation of RUNX)” strategy and RUNX depletion-induced activation of proapoptotic p53 in various cancers including MRT. Taken together, RUNX might be a potential and promising target for the treatment of MRT patients, and thus future clinical trials of our novel RUNX inhibitor Chb-M' are awaited.
We would like to thank Mr. Sohei Urakami for his donation to overcome the rare cancer.
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
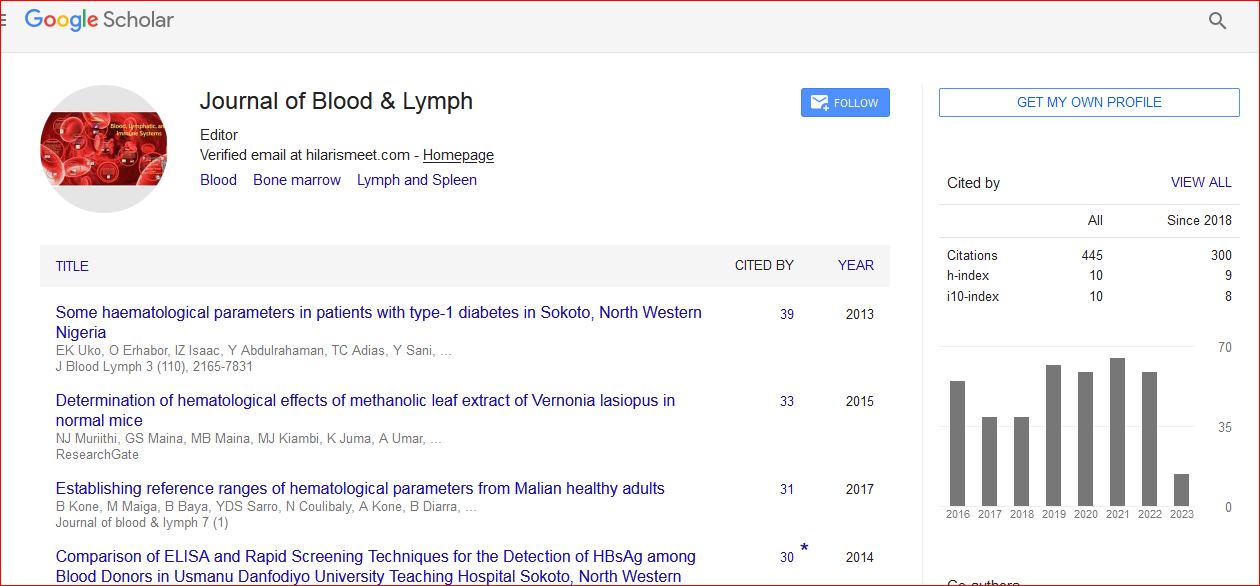
Journal of Blood & Lymph received 443 citations as per Google Scholar report