Commentary - (2025) Volume 15, Issue 1
Received: 15-Mar-2024, Manuscript No. JBL-24-129644;
Editor assigned: 18-Mar-2024, Pre QC No. JBL-24-129644 (PQ);
Reviewed: 03-Apr-2024, QC No. JBL-24-129644;
Revised: 15-Jan-2025, Manuscript No. JBL-24-129644 (R);
Published:
22-Jan-2025
, DOI: 10.37421/2165-7831.2025.15.341
Citation: Halteren, Astrid GS van. "Recurrent Somatic Mutations
in Pediatric Langerhans Cell Histiocytosis and their Significance in
Therapeutic Decision-Making." J Blood Lymph 15 (2025): 336.
Copyright: © 2025 Halteren AGSV. This is an open-access article distributed under the terms of the creative commons attribution license which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Langerhans Cell Histiocytosis (LCH) is one of the most frequently diagnosed histiocytic disorders in both adults and children [1]. Together with other types of non-Langerhans cell histiocytosis, LCH forms a spectrum of rare and clinically heterogeneous hematological disorders classified as histiocytic/dendritic cell neoplasms [2]. LCH manifests clinically either as a single or as multiple inflammatory lesions affecting virtually every organ system including the brain. The neoplastic features of its pathognomonic cells-CD1abrightCD207bright/ dim histiocytes-include clonal origin [3] and hyper activation of the Mitogen-Activated Protein Kinase (MAPK) pathway. The latter is evidenced by cytoplasmic expression of phosphorylated MEK and ERK proteins [4]. These two separately reported key findings propelled efforts by many histiocytosis research groups to find underlying somatic mutations [5]. As off 2010, a substantial number of mostly mutually exclusive somatic driver mutations have been identified. These mutations affect either components of the MAPK pathway itself [6-8] or other receptor tyrosine kinases which converge onto ERK [9,10].
The two most frequently affected genes in pediatric LCH are BRAF and MAP2K1 (MEK1), with mutational events occurring at hotspot locations in respectively exon 12 and exon 15 of the BRAF gen (chromosome 7) and in exon 2 or 3 of the MAP2K1 gene (chromosome 15). Notably, the most prevalent BRAF mutation- BRAFp. V600E-as well as recurrent MAP2K1 mutations have been found in non-Langerhans cell histiocytosis biopsies as well as in many other types of solid tumors. Hence, these pathological mutations are not uniquely associated with LCH presentation. Two recent studies have addressed the clinical associations of recurrent BRAF and MAP2K1 mutations in large, non-overlapping pediatric LCH cohorts. As the BRAFp.V600E frequency in our cohort nearly equaled its frequency in the French cohort, we virtually combined the two data sets to generate sufficient power to investigate the impact of the aforementioned hotspot mutations on LCH presentation and outcome. Either study individually, as well as the combined dataset, compellingly showed that BRAFp.V600E strongly associates with skin involvement and with disseminated disease affecting the liver, spleen and bone marrow. The latter clinical phenotype is exclusively seen in infants and is referred to as ‘high risk’ multisystem LCH, because of significant mortality risk if left untreated. Children with ‘high risk’ LCH less frequently presented with alternative BRAF or MAP2K1 mutations. Moreover, MAP2K1 mutations were more often associated with osseous lesions and BRAF exon 12 insertions or deletions with lung involvement. BRAFp.V600E seems also highly associated with neurodegeneration, a very rare and devastating late complication of LCH. This observation fits well with published data on post-mortal investigation of brain parenchyma tissue from a LCH patient who died with active neurodegeneration. The patient’s brain contained perivascular clusters of CD14+CD33+CD163+ myeloid/monocytic cells, which specifically stained with VE1 antibody identifying cytoplasmic BRAFV600E protein expression in formalin fixed paraffin embedded tissue. Hence, clinical heterogeneity in pediatric LCH seems to be driven, to a certain extent, by the type of driver mutation expressed by neoplastic lesional cells.
It has been known for almost two decades that children with disseminated LCH have an increased risk of forming new lesions (disease progression) under first line vinblastine and prednisone treatment. These observations formed the backbone of several consecutively performed studies initiated by the International Histiocyte Society. Importantly, these studies were completed at a time when lesional genotyping was not yet standard practice. The combined Dutch and French dataset confirmed that there is a higher need for second line chemotherapy in children with disseminated BRAFV600E+ LCH, as compared to similar patients with alternative mutations. Yet, lesional BRAFp.V600E status alone is not a prognostic factor for poor event-free survival independent of disease extent, a conclusion based on our observation that children with bone lesions expressing this mutation have comparable event-free survival as cases without this mutation. Since the current LCH-IV trial (NCT02205762) opened, several empiric studies reporting successful use of either vemurafenib or cobimetinib to treat refractory histiocytosis in respectively adults and children have been published. These so called inhibitors all target the derailed MAPK pathway by specifically acting on one of its components. Due to these promising observations, there is an increased interest in replacing second line chemotherapy by targeted therapy. Based on preliminary data, some pediatric oncologists even advocate that first line inhibitor treatment is a safe and sensible option, because it rapidly induces disease remission and may even prevent the development of permanent consequences like diabetes insipidus or neurodegeneration. In view of this discussion, there are some important facts to mention. First, results from the previous LCH-III clearly showed that the majority of children with disseminated LCH responded well to first line vinblastine and prednisone treatment and did not relapse after therapy completion. Since vemurafenib exposure can lead to up to grade 3 skin toxicity, it is questionable whether upfront use of targeted therapy is justified for all children with disseminated BRAFV600E+ LCH. There are two subgroups of patients who may, however, qualify for first line inhibitor therapy. Infants with ‘high risk’ LCH who either present with a >6 disease activity score or with a combination of LCH and hemophagocytic lymphohistiocytosis. Although both types of LCH presentation are rare, they are associated with increased mortality rates. These patients will likely benefit from upfront combination of chemotherapy and inhibitor therapy or from early switching to inhibitor therapy. The latter scenario should be followed by a second phase of chemotherapybased treatment to effectively eradicate the neoplastic stem/ progenitor cell that gives rise to neoplastic cells found in the patients’ blood and LCH lesions. The second exception where there seems consensus for upfront use of targeted therapy are children with clinically proven neurodegeneration. Either dabrafenib or tovorafenib (an oral, selective and central nervous system-penetrant type II RAF inhibitor currently in clinical development for various types tumors bearing activating RAF alterations) seem currently the best option for these patients. Dabrafenib combined with the MEK inhibitor trametinib is already registered for pediatric.
BRAFp.V600E+ low-grade glioma. In addition, tovorafenib was recently shown to be effective in relapsed/refractory pediatric lowgrade glioma. Of note, while several inhibitors have been approved by the Food and Drug Administration (FDA) as standard-of-care for adult histiocytosis patients, the European Medicines Agency (EMA) has not yet provided marketing authorization for their use in the context of histiocytic neoplasms. Hence, treatment of European pediatric patients has thus far been done only in off-label settings, as the two actively recruiting studies-COBRAH (2018-002222-23) and DRUP (NCT02925234) both enroll only histiocytosis patients >18 years of age. Prospective, multicenter studies combined with monitoring of mutation-bearing cells in the patients’ circulation are needed to address the success of either single or combined chemotherapy/targeted therapy protocols in pediatric LCH.
The histology data was kindly provided by Dr. R.E. Verdijk, Erasmus University Medical Center Rotterdam. I also want to thank Dr. C. van den Bos (Princess Maxima Center for Pediatric Oncology) for critical reading of the manuscript and Dr. R. Nell (Leiden University Medical Center) for his help with Figure.
Chromosomal location of the 3 most commonly detected driver mutations in pediatric Langerhans cell histiocytosis. (A) Visualization of intracellular signal transduction via the MAPK pathway and specific targeting of mutated BRAF protein by 3 different types of inhibitors mentioned in the main text. Histology shows neoplastic histiocytes in an orbital BRAF mutated LCH lesion, which stain positive for phosphorylated ERK protein as evidenced by the red color of their cytoplasm. (B) Schematic overview of the chromosomal locations of pediatric LCH-associated hotspot mutations in BRAF and MAP2K1. The most frequently affected exons are depicted in orange and the 3 most frequently found hotspot mutations are depicted on the right.
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
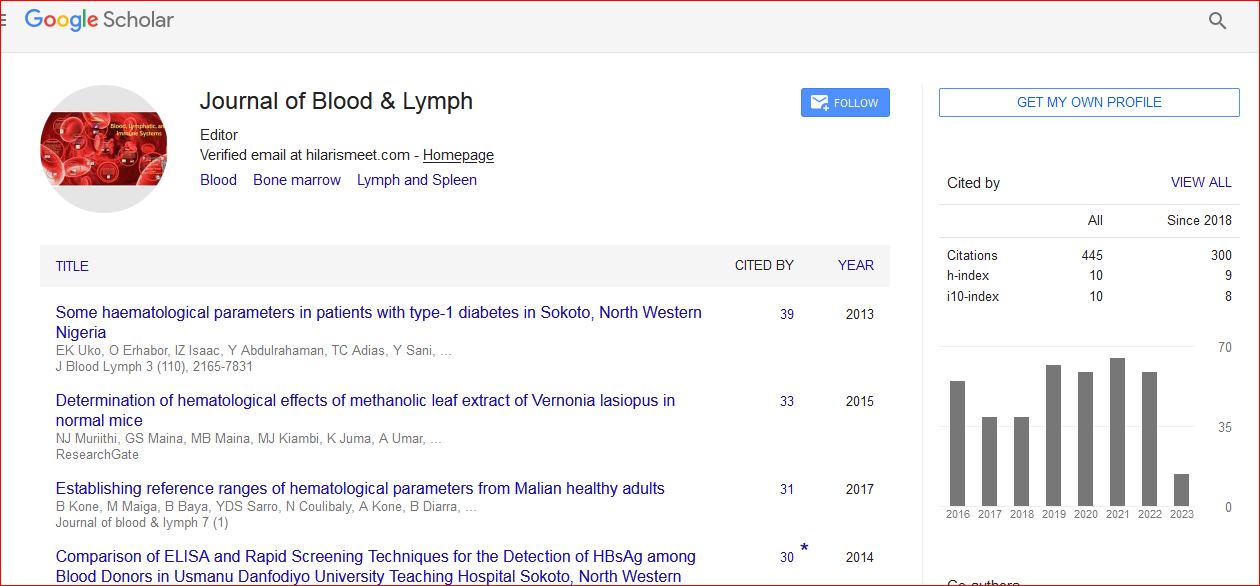
Journal of Blood & Lymph received 443 citations as per Google Scholar report