Perspective - (2023) Volume 12, Issue 3
Received: 01-May-2023, Manuscript No. jnc-23-106321;
Editor assigned: 03-May-2023, Pre QC No. P-106321;
Reviewed: 15-May-2023, QC No. Q-106321;
Revised: 22-May-2023, Manuscript No. R-106321;
Published:
31-May-2023
, DOI: 10.37421/2167-1168.2023.12.587
Citation: Kessler, Laurence. “Cystic Fibrosis: Challenges and Advances in Treatment.” J Nurs Care 12 (2023): 587.
Copyright: © 2023 Kessler L. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Cystic Fibrosis (CF) is a complex genetic disorder that affects thousands of individuals worldwide. It primarily impacts the lungs, digestive system and other organs, making it a life-altering condition. Although CF poses significant challenges to those living with it, advancements in research and treatment have improved the prognosis and quality of life for individuals with this condition. In this article, we will explore the causes, symptoms, diagnosis and recent breakthroughs in managing cystic fibrosis. Cystic fibrosis is caused by mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene, which regulates the movement of salt and water in and out of cells. These mutations lead to the production of a faulty CFTR protein or the absence of the protein altogether. As a result, the mucus in various organs becomes thick and sticky, causing blockages and impairing normal functions.
The symptoms of cystic fibrosis can vary widely depending on the severity of the disease and the organs affected. The most commonly observed signs and symptoms include persistent coughing, wheezing, recurrent lung infections (such as pneumonia and bronchitis), shortness of breath, poor growth or weight gain, salty-tasting skin, frequent bowel movements and digestive problems (such as malabsorption and pancreatic insufficiency). CF can also affect other organs like the liver, pancreas and reproductive system [1-3].
The symptoms of CF can vary widely from person to person, but they primarily affect the respiratory and digestive systems. Common symptoms include persistent coughing, wheezing, frequent lung infections, poor weight gain, salty-tasting skin, digestive issues and infertility in males. Since these symptoms are not exclusive to CF, diagnosis often involves a series of tests, including sweat tests, genetic testing, lung function tests and imaging studies [4].
The diagnosis of cystic fibrosis involves a combination of clinical evaluation, genetic testing and specialized diagnostic procedures. Newborn screening has proven to be crucial in detecting CF early, even before the onset of symptoms. A blood test checks for elevated levels of immunoreactive trypsinogen (IRT), followed by a genetic analysis to identify the specific CFTR gene mutations. Additional tests, including sweat chloride test, sputum culture, lung function tests and imaging studies, may be conducted to evaluate the extent of organ involvement and monitor disease progression [5].
Management and treatment
The management of cystic fibrosis involves a multidisciplinary approach that aims to control symptoms, prevent complications and improve overall wellbeing. Treatment plans are tailored to the individual's specific needs and may include:
Airway clearance techniques: These techniques, such as chest physiotherapy and the use of vibrating vests, help loosen and remove mucus from the lungs, reducing the risk of infections.
Medications: Various medications are used to manage CF symptoms. Bronchodilators help open the airways, antibiotics treat lung infections, mucus-thinning agents improve mucus clearance and pancreatic enzyme supplements aid digestion.
Nutritional support: Individuals with CF often require a high-calorie diet to maintain proper nutrition. Dietitians work closely with patients to develop meal plans that meet their nutritional needs.
Lung transplantation: In severe cases, where lung function significantly declines, a lung transplant may be considered. This procedure can improve quality of life and increase life expectancy for eligible candidates.
While there is currently no cure for cystic fibrosis, advancements in medical research and treatment have significantly improved the quality of life and life expectancy for individuals with CF. The management of CF typically involves a multidisciplinary approach, including medical interventions, physical therapy, nutritional support and psychological care. Medications such as bronchodilators, mucus-thinning drugs, antibiotics and pancreatic enzyme replacement therapy (PERT) are commonly prescribed to alleviate symptoms and manage complications. Additionally, lung transplantation may be considered in severe cases where lung function declines significantly.
Recent breakthroughs
In recent years, significant progress has been made in the treatment of cystic fibrosis, thanks to advancements in understanding the disease at a molecular level. One notable breakthrough is the development of CFTR modulator drugs. These medications, such as ivacaftor, lumacaftor, tezacaftor and elexacaftor, target specific CFTR mutations and help restore their function. This novel approach has shown remarkable results in improving lung function, reducing hospitalizations and enhancing overall health for individuals with specific CF mutations.
Furthermore, gene therapy and CRISPR-Cas9 technology hold promise for future treatments. Gene therapy involves introducing a functional CFTR gene into the cells of individuals with CF, while CRISPR-Cas9 enables the correction of defective CFTR genes directly. Although these approaches are still in early stages of development, they offer hope for potential cures or longterm treatments.
Support and research
Living with cystic fibrosis can be physically and emotionally challenging, not only for individuals with the condition but also for their families. Support from healthcare professionals, support groups and organizations dedicated to CF can provide valuable resources, education and a sense of community for those affected.
Research efforts continue to expand our understanding of cystic fibrosis and improve treatment options. Scientists are investigating novel therapies, exploring personalized medicine approaches and working towards better outcomes for all individuals with CF.
Emerging therapies and research
Recent breakthroughs in CF research have led to the development of innovative therapies targeting the underlying genetic defects. One such breakthrough is the advent of CFTR modulator drugs, which aim to correct the CFTR protein's function and improve the transport of salt and water across cell membranes. These modulators have shown promising results in specific CF mutations, significantly improving lung function and reducing exacerbations. Ongoing research is focused on expanding the availability of these therapies to a broader range of CF mutations and developing new treatment approaches.
Cystic fibrosis remains a complex condition, but advancements in research and treatment have transformed the lives of individuals with CF. From improved symptom management to the development of CFTR modulator drugs, there is reason to be optimistic about the future. Ongoing research and support from the medical community, combined with a multidisciplinary approach to care, provide hope for continued progress in the understanding and treatment of cystic fibrosis.
None.
No conflict of interest.
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
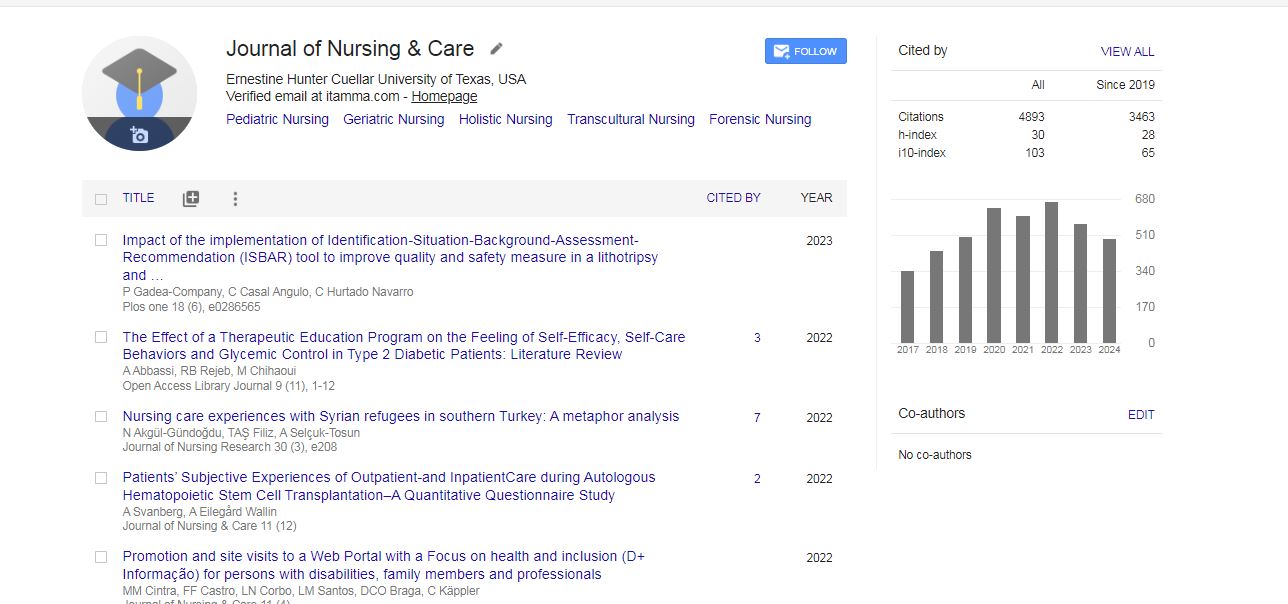
Journal of Nursing & Care received 4230 citations as per Google Scholar report