Ezeldeen Alsorori, Carmen Cantisani, Ahmad Faiq Naqeshbandi, Mohamad Goldust, Salvatore Lampitelli,Franca Cantoresi
U.O.C Dermatologia, Dipartimento di scienze mediche, Policlinico Umberto , Italia
Mazandaran University of medical sciences, Iran
University of Science and technology hospital Sanaa, Yemen
Posters & Accepted Abstracts: J Cosmo Trichol
Primary immunodeficiencies(PIDS) are rare,inherited diseases, characterized by altered function or absence of immune cells. Among them is leukocyte adhesion deficiency type I (LAD-I), an autosomal recessive disorder characterized by primary immunodeficiency, caused by mutations in the ITGB2 gene which produces inability of leucocytes to migrate towards the area of inflammation and is associated with recurrent life-threatening bacterial and fungal infections. Pyoderma gangrenosum(PG) is an uncommon noninfectious neutrophilic dermatosis, characterized by recurrent, necrotic ulcers. It is a diagnosis of exclusion and can be challenging and its management is empirical, with local(topical tacrolimus or intralesional triamcinolone) or systemic immunosuppressive therapy (oral or intravenous glucocorticoids, sulfasalazine, especially in cases associated with crohn�??s disease, cyclosporine and, recently, anti-TNF drugs such as Infliximab, Etanercept, Adalimumab). Though skin ulcerations are common, predominant clinical presentation as PG can often mimic other diseases. It is unusual in children even more in LAD-I. Here we present a Yemenian family with LAD-I from consanguineous relatives. All patients had history of chronic recurrent skin ulcerations without any bleeding tendency, associated with persistent neutrophilia and requiring steroids and antibiotics. There was no history of delayed cord separation and the condition was initially diagnosed as epidermolysis bullosa, but successively as PG. LAD-I should be kept in mind while evaluating patients with PG especially in children with persistent neutrophilia in the absence of other rheumatological disorders. Its diagnosis is extremely important from the management perspective, as treating these patients without adequate antibiotic cover may be fatal, as happened to one of our patient, and these patients often require hematopoietic stem cell transplantation for permanent cure. Therefore, genetic counseling especially in population with high consanguinity is mandatory.
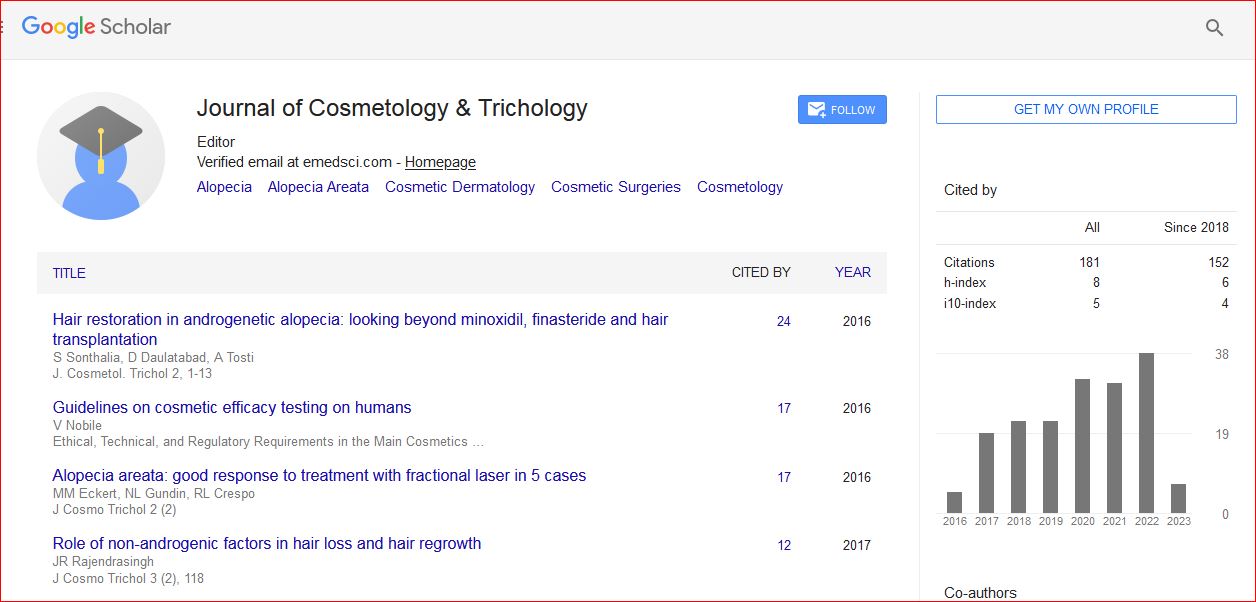
Journal of Cosmetology & Trichology received 180 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 