Short Communication - (2025) Volume 9, Issue 3
Received: 01-May-2025, Manuscript No. hps-26-184440;
Editor assigned: 05-May-2025, Pre QC No. P-184440;
Reviewed: 19-May-2025, QC No. Q-184440;
Revised: 22-May-2025, Manuscript No. R-184440;
Published:
29-May-2025
, DOI: 10.37421/2573-4563.2025.9.351
Citation: Farouk, Ahmed R.. ”Preserving Beta Cell Function: A Diabetic Pathogenesis.” J Hepatol Pancreat Sci 09 (2025):351.
Copyright: © 2025 Farouk R. Ahmed This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use,distribution and reproduction in any medium, provided the original author and source are credited.
Pancreatic beta cell dysfunction is a fundamental aspect of the pathological processes underlying both type 1 and type 2 diabetes mellitus. This cellular malfunction is characterized by diminished insulin synthesis, impaired insulin secretion, and an elevated rate of apoptosis, contributing significantly to the disease state [1].
Inflammation plays a pivotal role in the attrition and compromised function of beta cells within the islets of Langerhans. Pro-inflammatory cytokines, such as tumor necrosis factor-alpha (TNF-α) and interleukin-1 beta (IL-1β), have been observed to directly impede insulin secretion and foster beta cell demise [2].
Endoplasmic reticulum (ER) stress emerges as a critical determinant in beta cell failure associated with diabetes. Dysregulation of ER homeostasis, often triggered by hyperglycemia and elevated free fatty acids, initiates the unfolded protein response (UPR). While the UPR can offer transient adaptive benefits, prolonged ER stress ultimately activates apoptotic cascades, leading to beta cell loss [3].
Accumulation of lipids and the ensuing lipotoxicity represent significant contributors to beta cell dysfunction, particularly in the context of type 2 diabetes. Excessive free fatty acids can compromise glucose-stimulated insulin secretion and promote apoptosis through mechanisms involving ER stress and oxidative damage [4].
Glucotoxicity, a consequence of sustained hyperglycemia, directly inflicts damage upon beta cells. This involves alterations in gene expression, exacerbation of oxidative stress, and disruption of insulin synthesis and secretion processes [5].
Emerging therapeutic modalities targeting the incretin system, including glucagon-like peptide-1 (GLP-1) receptor agonists and dipeptidyl peptidase-4 (DPP-4) inhibitors, have transformed type 2 diabetes management by augmenting glucose-dependent insulin release and curbing glucagon secretion [6].
Sodium-glucose cotransporter-2 (SGLT2) inhibitors, initially conceived for glycemic control, have demonstrated multifaceted benefits extending beyond glucose lowering, notably in cardiovascular and renal health. Their precise influence on beta cell well-being is an active area of investigation [7].
The identification of beta cell regenerative potential marks a significant breakthrough in diabetes research. Strategies aimed at stimulating beta cell proliferation and differentiation from progenitor cells are under active exploration, offering hope for restoring functional beta cell mass [8].
Oxidative stress contributes detrimentally to beta cell dysfunction and apoptosis by damaging critical cellular components and interfering with insulin signaling pathways. Antioxidant interventions are being pursued to counteract this cellular damage [9].
Epigenetic modifications, encompassing DNA methylation and histone alterations, are increasingly recognized for their regulatory influence on beta cell function and their involvement in diabetes pathogenesis. These changes can impact genes vital for insulin production, secretion, and beta cell viability [10].
Pancreatic beta cell dysfunction is central to the pathogenesis of both type 1 and type 2 diabetes. This dysfunction involves impaired insulin synthesis, secretion, and increased apoptosis. Mechanistically, factors like glucotoxicity, lipotoxicity, inflammation, and endoplasmic reticulum stress play significant roles in beta cell failure [1].
Inflammation is a critical driver of beta cell loss and dysfunction in diabetes. Pro-inflammatory cytokines, such as TNF-α and IL-1β, directly impair insulin secretion and promote beta cell apoptosis. Targeting these inflammatory pathways offers a promising avenue for preserving beta cell mass and function [2].
Endoplasmic reticulum (ER) stress is a significant contributor to beta cell failure in diabetes. Conditions like hyperglycemia and elevated free fatty acids disrupt ER homeostasis, leading to the unfolded protein response (UPR). While the UPR can initially be adaptive, chronic ER stress triggers apoptotic pathways, ultimately causing beta cell demise [3].
Lipid accumulation and lipotoxicity are key culprits in beta cell dysfunction, particularly in type 2 diabetes. Excessive free fatty acids can impair glucose-stimulated insulin secretion and promote apoptosis through various mechanisms, including ER stress and oxidative stress [4].
Glucotoxicity, arising from chronic hyperglycemia, directly damages beta cells by altering gene expression, promoting oxidative stress, and impairing insulin synthesis and secretion. Controlling blood glucose levels through conventional and novel antidiabetic agents remains a cornerstone of preventing beta cell decline [5].
Incretin-based therapies, including GLP-1 receptor agonists and DPP-4 inhibitors, have revolutionized type 2 diabetes management by enhancing glucose-dependent insulin secretion and suppressing glucagon release. These agents also offer beta cell protective effects, potentially through improved beta cell mass and function [6].
Sodium-glucose cotransporter-2 (SGLT2) inhibitors, initially developed for glycemic control, have demonstrated significant cardiovascular and renal benefits beyond their glucose-lowering effects. Their impact on beta cell health is an area of ongoing research, with some evidence suggesting indirect protection through improved metabolic profiles and reduced glucotoxicity [7].
The discovery of beta cell regenerative potential is a significant advance. Strategies aimed at promoting beta cell proliferation and differentiation from progenitor cells are being investigated. This includes exploring growth factors, signaling pathways, and stem cell-based therapies [8].
Oxidative stress plays a detrimental role in beta cell dysfunction and apoptosis by damaging cellular components and impairing insulin signaling. Antioxidant strategies, both dietary and pharmacological, are being explored to mitigate this damage. Understanding the specific sources and pathways of oxidative stress in the diabetic islet is crucial for developing targeted interventions [9].
Epigenetic modifications, including DNA methylation and histone modifications, are increasingly recognized as important regulators of beta cell function and are implicated in the pathogenesis of diabetes. These alterations can affect the expression of genes critical for insulin production, secretion, and beta cell survival [10].
Pancreatic beta cell dysfunction is central to diabetes pathogenesis, involving impaired insulin synthesis, secretion, and increased apoptosis. Key mechanisms contributing to beta cell failure include glucotoxicity, lipotoxicity, inflammation, and endoplasmic reticulum stress. Current and emerging therapies focus on protecting beta cells, restoring function, and promoting regeneration. These include incretin-based therapies and SGLT2 inhibitors, which offer benefits beyond glucose lowering. Research into beta cell regeneration, antioxidant strategies, and epigenetic modifications presents novel avenues for therapeutic intervention. Understanding the complex interplay of these factors is crucial for developing effective treatments to preserve beta cell mass and function in diabetic individuals.
None
None
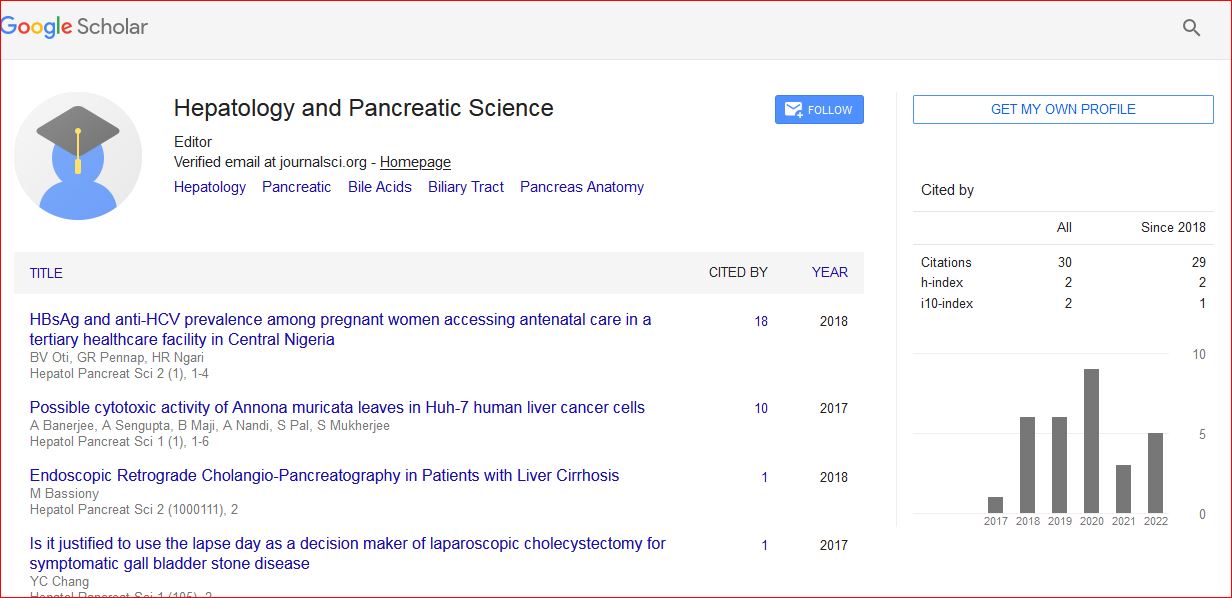
Hepatology and Pancreatic Science received 34 citations as per Google Scholar report