Mini Review - (2020) Volume 9, Issue 4
Received: 12-Sep-2020
Published:
25-Sep-2020
, DOI: 10.37421/2168-9547.2020.09.245
Citation: Sudhansu Sekhar Patra. Genomics of Human and Animal Populations. Mol Biol 9 (2020):245. doi: 10.37421/mbl.2020.09.245
Copyright: © 2020 Sudhansu Sekhar Patra. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Genetic variation inspirations genetic factor expression, and this type of variation in gene expression may be professionally mapped to exact genomic regions and variants. Now we have used gene expression profiling of Epstein-Barr virus–transformed lymphoblastoid cell lines of all 270-separate genotyped in the HapMap Grouping to explain the thorough features of genetic variation fundamental gene expression variation. We discovery that gene expression is transmissible and that difference between populations is in agreement by earlier small-scale studies. A full connotation analysis of over 2.2 million common SNPs per population (5% frequency in HapMap) by gene expression recognized at least 1,348 genes with association signals in cis and at least 180 in trans. Replication on minimum one independent population was achieved for 37% of cis signals and 15% of trans signals, correspondingly. Our results powerfully support a profusion of cis-regulatory difference in the human genome. Detection of trans effects is limited but proposes that regulatory variation might be the key main effect contributing to phenotypic variation in humans. We also discover several methodologies that advance the present state of analysis of gene expression variation.
Gene, Genomics, Animal, Human, Populations.
Recent developments in genotyping mechanism have facilitated genomewide scans for natural collection. Detecting marks of natural collection sheds light on human evolution and it could help to recognize genetic variants that influence standard human phenotypic variation as well as virus susceptibility. Here we are mainly focus on studies of natural collection in modern humans who originated ∼200,000 years ago in Africa and it’s migrated across the globe ∼50,000–100,000 years ago. Movement into new environments, as well as variations in culture and technology, counting plant and animal domestication, resulted in resident adaptation to diverse environments. We abridge statistical methods for detecting marks of natural collection and for the distinguishing effects of demographic history from natural selection. On a genome-wide scale, immune-related genes are main targets of positive collection. Genes related with reproduction and fertility also are fast evolving. Further samples of current human adaptation add genes connected with lactase persistence, eccrine glands, and response to hypoxia. Lastly, we emphasize the essential to supplement scans of collection with useful studies to demonstrate the physiologic impact of candidate loci.
Animal coloration has been traditionally the mark of genetic and evolutionary studies. However, until very currently, the study of the genetic base of animal coloration has been mostly limited to model species, whereas research on non-model species has remained either ignored or mostly based on applicant approaches, and thereby incomplete by the knowledge found in model species. Current high-throughput sequencing machineries allow us to stunned previous limits, and open new streets to study the genetic foundation of animal coloration in a larger number of species and colour traits, and to speech the general relevance of dissimilar genetic structures and their inferences for the evolution of colour. In this type of review, we highlight features where genome-wide studies might be of main utility to fill in the gaps in our sympathetic of the biology and evolution of animal coloration. The new genomic methods have been punctually accepted to study animal coloration though considerable work is still wanted to consider a bigger range of species and colour traits, such as those exhibiting incessant variation or based on reflective structures. We claim that a robust progression in the study of animal coloration will also need large efforts to validate the functional role of the genes and variants exposed using genome-wide tools.
The study of animal coloration has been crucial for the growth of biological sciences, principally for the fields of genetics and evolutionary biology. Pioneering geneticists like Morgan, Bateson or Haldane studied the inheritance of colour traits to found the fundamentals of Mendelian genetics. Similarly, studies of coloration in wild populations, which was Kettlewell's studies on the act of natural collection on the black and pale morphs of the sprayed moth, Biston betularia, or Endler's studies on the joint part of sexual and natural collection on elucidation colour variation in guppies, Poecilia reticulata, have also moulded our considerate of evolution. For practical motives, many of these studies took benefit of colour traits exhibiting relatively modest separate variation and inheritance patterns. Also, for applied explanations, research on animal coloration has continued and flourished frequently on the model systems used through these pioneers (Drosophila and mice, for instance), by only a limited new species existence accepted as model systems later on (e.g. the zebrafish, Danio rerio. Our present considerate of the genetics of animal coloration is therefore inadequate to a handful of well-studied systems, and does not account for the extant variation of life forms and colour traits. In natural situations, separate variation is the exemption rather than the rule. Most colour traits vary incessantly between two extreme values and are complexly based in the confession of numerous pigments and/or on the spatial preparation of integumentary structures. The appearance of these traits can be powerfully strongminded by genetic factors but also by the environment or by the communication of genetic and environmental factors. Finally, colour traits can have dissimilar types of roles and adaptive functions, which can result in dissimilar evolutionary histories and original genetic architectures, even when seeing colour traits of high similarity.
Remarkable exclusions aside (e.g. mapping studies in Heliconus, cichlids and white-throated sparrows, Zonotrichia albicollis, classifying the fundamental genes and genetic structures of such a diverse spectrum of colour traits has been established little attention in the wild, partly because previous methods to find loci answerable for colour variation were difficult, exclusive and/or unfeasible in natural populations. In current years and cheers to the seminal work of N. Mundy and H. Hoekstra on the melanocortin-receptor 1, MC1R, unscrambling the genetic basis of coloration has mostly founded on the more accessible candidate-gene approach. This method measures the character that genes recognized to control coloration on model species play in non-model species. It has been established very valuable to achieve a better considerate of colour variation in a wide range of species, importance the recurrent role of certain colour genes, like the MC1R, in mediating adaptive colour changes in several species.
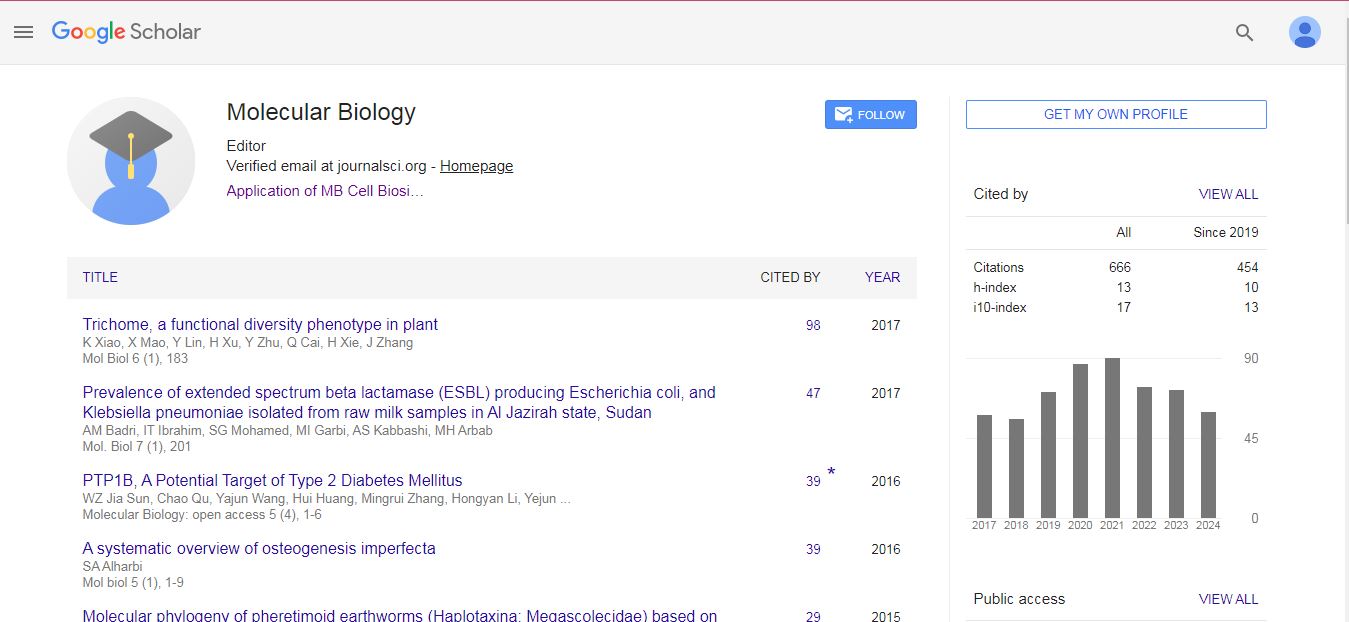
Molecular Biology: Open Access received 607 citations as per Google Scholar report