Mini Review - (2022) Volume 8, Issue 4
Received: 24-Sep-2022, Manuscript No. ELJ-22-75867;
Editor assigned: 27-Sep-2022, Pre QC No. P-75867;
Reviewed: 07-Oct-2022, QC No. Q-75867;
Revised: 12-Oct-2022, Manuscript No. R-75867;
Published:
18-Oct-2022
, DOI: 10.37421/2472-0895.2022.8.169
Citation: Cristiana, Eva. “Examining New Cases Clinically and Genetically.” Epilepsy J 8 (2022): 169.
Copyright: © 2022 Cristiana E. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
The rare disorder Wolf-Hirschhorn syndrome (WHS), which is caused by a distal 4p deletion, is characterised by craniofacial dysmorphism, congenital fusion abnormalities, hypotonia, intellectual impairment, and epilepsy. The clinical characteristics depend on the magnitude of the deletion. Our goals included identifying unusual distinct traits in a cohort of seven patients with 4p deletion and evaluating the usefulness of Multiplex ligation-dependent probe amplification (MLPA) (cheap and sensitive test)-combined kits as a diagnostic test and tool for cases that need additional research (chromosomal microarray analysis-CMA, karyotype). The basic characteristics of facial dysmorphism, intellectual disability, postnatal development delay, heart abnormalities, and hypotonia were detected during a clinical examination for all cases. We occasionally noticed renal anomalies, immunodeficiencies, convulsions, and structural brain abnormalities. A relatively limited number of cases of prenatal growth retardation were found, however postnatal growth failure was always present. In each case, karyotype and/or MLPA genetic testing supported the clinical diagnosis. In conclusion, it is important to look for the unusual signs of immunodeficiency, renal, and brain abnormalities. Although CMA is the industry standard test, in our experience, MLPA is also a trustworthy screening approach because the instances that were detected were either validated by MLPA or chosen for further research.
Craniofacial dysmorphism • Growth retardation • IUGR
The distal region of the short arm of chromosome 4 is absent in Wolf- Hirschhorn syndrome (WHS), a rare contiguous gene deletion syndrome with an incidence of 1:20,000–50,000 births and a male to female ratio of 2:1 [1,2]. Intrauterine growth retardation, followed by small stature, low weight, hypotonia, intellectual disability, epilepsy, and a particular type of craniofacial dysmorphism ("Greek warrior helmet"), are characteristics of the WHS phenotype. Skeletal anomalies, congenital heart defects, eye abnormalities, hearing loss, genitourinary tract issues, and immunological disorders are further related clinical symptoms [3]. Wide nasal bridge that extends to the forehead, high anterior hairline with prominent glabella, sharply arched eyebrows, widely separated eyes, epicanthus, short philtrum, downturned corners of the mouth, and micrognathia are some suggestive craniofacial traits. The majority of patients have low-set or posteriorly angulated ears with pits or tags, as well as microcephaly and poorly developed ears (lobeless pinnae, underdeveloped or missing cartilage) [3]. The distal short arm of chromosome 4 is typically partially deleted, however the WHS phenotype can also result from complicated chromosomal rearrangements, such as translocations or ring chromosomes. De novo or inherited from a parent with a balanced rearrangement, the unbalanced translocations can occur. The most common translocations are: (1) t(4p;8p rearrangements, but t(4p;7p), t(4p;11p), t(4p;20q), t(4p;21q), and t(4p;12p), (2) inverted duplications linked to terminal deletions on the same 4p arm, or (3) imbalanced pericentric inversions [3,4]. The size of the deletion varies significantly, ranging from less than 2 Megabases to 30 Mb. Based on the amount of the 4p deletion, there are three different types of WHS phenotypes: a modest deletion (not more than 3.5 Mb), which is typically linked to a moderate phenotype (clinical manifestations are limited to the typical facial dysmorphism, growth delay, mild intellectual disability, and seizures).
The very large deletion (exceeding 22-25 Mb) has the most severe phenotype and is frequently difficult to identify as WHS, whereas the large deletion is the most common and is associated with more severe manifestations, such as hypotonia, severe growth delay, significant neurodevelopmental impairment, and major malformations on top of those from the mild type [4]. The two most significant predictive indicators for neurodevelopmental difficulties are the genetic abnormality and the pattern of seizures [4]. Approximately 2 Mb from the telomere 4p, in band 4p16.3 [5] (a region of 200-750 kb rich in genes- WHSC1, LETM1, NSD2, CPLX1, PIGG being usually deleted) [6,7], is where the WHS minimal critical region (MCR) is found. Clinical examination and genetic testing are required for the diagnosis. Karyotypic analysis can identify deletions larger than 5 MB, but molecular diagnostic methods such as CMA, MLPA, and fluorescence in situ hybridization (FISH) are required for smaller deletions (microdeletions). About 50-60% of the deletions may be found using conventional and high resolution chromosomal analysis, but more than 95% can be found utilising FISH and a WHSCR probe. All known deletions of the WHSCR may be found using chromosomal microarray (CMA), which can also tell whether a deletion is pure or a result of a more complicated rearrangement. The CMA may be accompanied by conventional cytogenetics to characterise any complicated abnormality that may be present. A multiplex PCR technique called MLPA uses up to 40 probes, each of which is tailored to a particular DNA sequence [8]. The use of MLPA in genetic testing labs for the molecular diagnosis of various disorders has increased significantly in recent years. MLPA is a high throughput assay that enables thorough identification of gene doses with very inexpensive tools and materials. Microdeletions that may not be detected by FISH can be found with a combined MLPA test (combination of particular kits for microdeletion screening, followed up by specific kits for microdeletion confirmation). Given that WHS has a high mortality rate (30% within the first two years of life), supportive management may be helpful as there are no particular treatments for the condition. Infections of the respiratory tract and congenital cardiac conditions are the most frequent complications that result in death [1].
Our work sought to establish MLPA as a valid technique for the diagnosis of WHS by identifying uncommon unique characteristics of WHS, exploring whether there are significant changes based on deletion size and whether there is a relationship between deletion size and phenotypic severity.
Case 1
A 13-year-old boy in Case 1 is the first child of a young, unrelated, seemingly healthy couple; there are no other cases in the family. The pregnancy went without a hitch, and a corpus callosum agenesis was discovered during a foetal ultrasound taken during 36 weeks of amenorrhea (WA). The baby was born by caesarean section at full term, weighed 3600 g, stood 52 cm tall, and had an occipital-frontal circumference of 36.5 cm. The baby also received an Apgar score of 8. The patient suffers recurrent respiratory infections, and postnatal development was severely delayed (raised head at 18 months (Mo), sat without support at 2 years (Y), and no speech at age 13 Y). At the age of 13 years and 7 months, physical examination results (Wt: -0.89 SD, Ht: -2.39 SD, OFC: -1.23 SD) showed that the patient had a dysmorphic face, a lacrimal system defect, a small palate defect, pectus carinatum, diastasis recti, hypospadias, undescended testes, a sacral sinus, delayed tooth eruption, spastic quadriplegia Agenesis of the corpus callosum (ACC) and slight ventricular system enlargement were also visible on MRI. Bilateral ocular atrophy and heart abnormalities, such as an atrial septal defect (ASD), were also discovered in this case. A 2 Mb deletion in the WHS area was verified by XY and MLPA; the karyotype was 46.
Case 2
The lone child of unrelated parents in the second case is a 4-year-old girl. The pregnancy progressed to polyhydramnios and a potentially dangerous miscarriage. The baby was delivered via caesarean section at term (Apgar score of 9, weight 2,500 g, height 48 cm, and OFC 31.5 cm). Development after birth was postponed. There have been reports of Jacksonian seizures in the right side of the body. Small size (Wt: -3.2 SD, Ht: -4.15 SD), microcephaly (OFC: -4.66 SD), dysmorphic face, congenital cataract, dental defects, sacral sinus, two tuberous hemangiomas, and hypotonia were discovered during the physical examination. Additionally, the patient has a patent ductus arteriosus, ventricular septal defect, and ASD (PDA). A 2 Mb deletion in the WHS area was confirmed by MLPA P096 Probemix, and the karyotype was 46,XX.
Case 3
A 2-year-old kid in Case 3 is the second child of an unrelated, seemingly healthy couple. There are no other cases in the family. With intrauterine growth retardation, pregnancy evolved. A caesarean surgery was used to deliver the infant at 36 weeks (Wt: 1400 g; Ht: 30 cm; OFC: 28 cm). Development after birth is slowed. The most recent assessment (1 year and 6 months) showed small size (Wt: -7.09 SD, Ht: -5.7 SD), microcephaly (OFC: -4.81 SD), and a recognisable dysmorphic face.
In addition to the deletion's high degree of heterogeneity in terms of size and etiological mechanism, Wolf-Hirschhorn syndrome also exhibits high levels of clinical variability. Our chosen examples effectively demonstrate this. The majority of WHS patients (55%) had a "pure" deletion with no further cytogenetic abnormalities; the remainder had a more complex cytogenetic profile, including derived chromosome 4 from an imbalanced translocation, ring 4 chromosome, or a 4p-mosaicism.
The condition in cases 1, 2, and 3 was brought on by microdeletions (2 Mb) that affect the crucial part of 4p16.3 and cannot be seen on the karyotype. After the parents' cytogenetic analysis, the father of patient 4 was revealed to have a balanced translocation between chromosomes 4 and 17 (46,XY,1qh+,t(4;17) (p15.33;p13.3)). This was not unexpected because a carrier of balanced translocation produces a mis-segregation of derived chromosomes during meiosis in about 15% of instances with WHS. Chromosome 8p translocations are the most frequent in WHS, closely followed by translocations involving chromosomes 7p, 11p, and 12p. Due to the trisomy material's alteration of the phenotypic, patients with imbalanced translocations typically exhibit some divergence from conventional clinical symptoms.
A rare genetic condition known as Wolf-Hirschhorn syndrome has severe prognoses and varied clinical symptoms. Although the syndrome is commonly suggested by the general clinical features, it can be challenging to diagnose because of the vast range of symptoms. Our cases highlight the wide range of WHS: hearing loss is also linked to significant deletions, while facial dysmorphism and developmental delay are also frequent. Renal and brain problems should also be suspected. Therefore, in these patients, cytogenetic and molecular studies are crucial. Despite the fact that 85% of instances include de novo mutations, derivative chromosomes produced by a balanced translocation that existed in one of the parents were discovered in 15% of cases. Therefore, other genetic tests (CMA, MLPA, FISH, and karyotype) are required to complete the diagnosis in patients with WHS, and the chromosomal analysis of the parents is now required to determine the likelihood that there will be another case in the family. Although CMA is the gold standard test for WHS diagnosis, MLPA may be used as a screening approach before doing CMA in some circumstances depending on the capabilities of the laboratory.
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
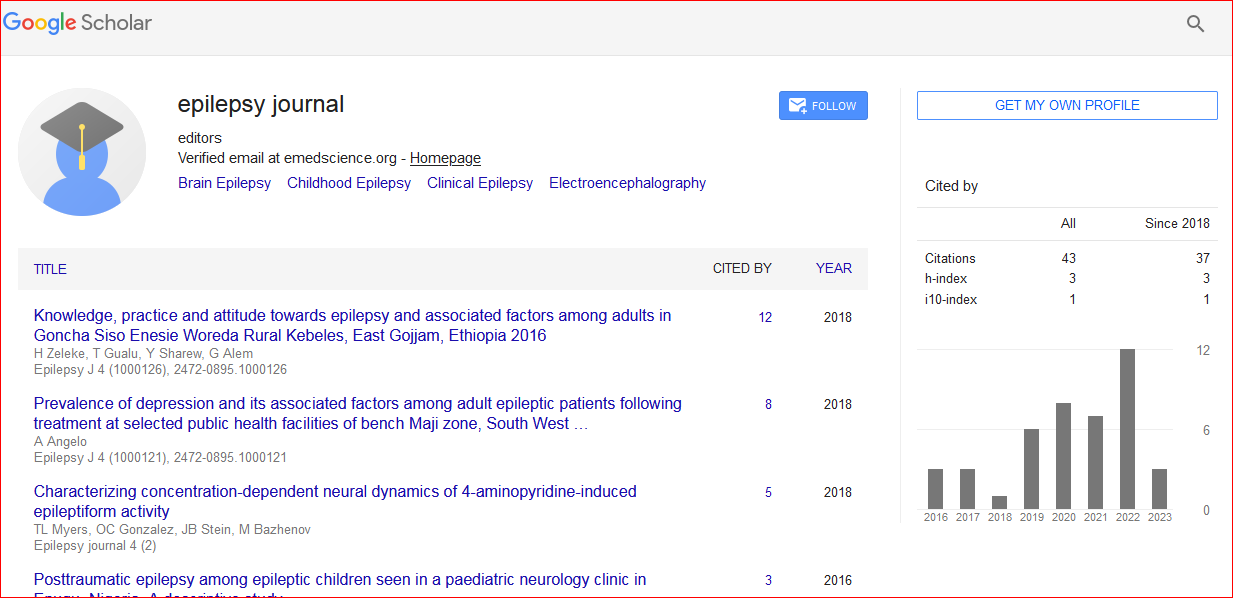
Epilepsy Journal received 41 citations as per Google Scholar report