Mohammad Reza Rafati and Ehsan Yousefi Mazhin*
DOI: 10.37421/ 2472-0895.2022.8.166
Valproate is an antiepileptic drug which is commonly used for the treatment of focal and/or generalized epilepsy and mood disorders. We have reported a case of 10-years-old, 30 kg boy was admitted in the pediatric intensive care unit due to status epilepticus and then was received some antiepileptic drugs including valproate. Almost two weeks after starting valproate, the total serum level of valproate was measured that was very low. We investigated the cause of low valproate levels in this patient.
DOI: 10.37421/2472-0895.2022.8.170
Extreme cases of intractable childhood epilepsy might culminate in an illness called epileptic encephalopathy. Along with being fatal in certain instances, the illness can result in significant delays in the development of cognitive, sensory, and motor functions. Early infantile SCN8A encephalopathy is associated with missense mutations in SCN8A, which encodes Nav1.6, a key subunit of the voltage-gated sodium channel in neurons and muscles. In this case report, we describe a 5-month-old child who has a new missense mutation associated with SCN8A encephalopathy. The findings of blood and metabolic testing, electroencephalogram (EEG), and brain magnetic resonance imaging (MRI), with the exception of uncontrollable seizures and autistic characteristics, were all normal. Genetic sequencing should be taken into consideration to determine the underlying genetic origins of these mutations since the phenotypes brought on by these mutations cannot be distinguished by any clinical, neuroimaging, or electrophysiological criteria. The administration of oxcarbazepine, as opposed to phenytoin, which is advised as a lastresort treatment for SCN8A encephalopathy, reduced this patient's uncontrollable seizures.
DOI: 10.37421/2472-0895.2022.8.169
The rare disorder Wolf-Hirschhorn syndrome (WHS), which is caused by a distal 4p deletion, is characterised by craniofacial dysmorphism, congenital fusion abnormalities, hypotonia, intellectual impairment, and epilepsy. The clinical characteristics depend on the magnitude of the deletion. Our goals included identifying unusual distinct traits in a cohort of seven patients with 4p deletion and evaluating the usefulness of Multiplex ligation-dependent probe amplification (MLPA) (cheap and sensitive test)-combined kits as a diagnostic test and tool for cases that need additional research (chromosomal microarray analysis-CMA, karyotype). The basic characteristics of facial dysmorphism, intellectual disability, postnatal development delay, heart abnormalities, and hypotonia were detected during a clinical examination for all cases. We occasionally noticed renal anomalies, immunodeficiencies, convulsions, and structural brain abnormalities. A relatively limited number of cases of prenatal growth retardation were found, however postnatal growth failure was always present. In each case, karyotype and/or MLPA genetic testing supported the clinical diagnosis. In conclusion, it is important to look for the unusual signs of immunodeficiency, renal, and brain abnormalities. Although CMA is the industry standard test, in our experience, MLPA is also a trustworthy screening approach because the instances that were detected were either validated by MLPA or chosen for further research.
DOI: 10.37421/2472-0895.2022.8.168
Quantitative electroencephalography (QEEG) is becoming an increasingly common method of diagnosing neurological disorders and, following the recommendations of The American Academy of Neurology (AAN) and the American Clinical Neurophysiology Society (ACNS), it can be used as a complementary method in the diagnosis of epilepsy, vascular diseases, dementia, and encephalopathy. However, few studies are confirming the importance of QEEG in the diagnosis of mental disorders and changes occurring as a result of therapy; hence, there is a need for analyses in this area. The aim of the study is analysis of the usefulness of QEEG in the diagnosis of people with generalized anxiety disorders. Our research takes the form of case studies. The paper presents an in-depth analysis of the QEEG results of five recently studied people with a psychiatric diagnosis: generalized anxiety disorder. The results show specific pattern amplitudes at C3 and C4. In all of the examined patients, two dependencies are repeated: low contribution of the sensorimotor rhythm (SMR) wave amplitudes and high beta2 wave amplitudes, higher or equal to the alpha amplitudes. The QEEG study provides important information about the specificity of brain waves of people with generalized anxiety disorder; therefore, it enables the preliminary and quick diagnosis of dysfunction. It is also possible to monitor changes due to QEEG, occurring as a result of psychotherapy, pharmacological therapy and EEG-biofeedback.
DOI: 10.37421/2472-0895.2022.8.167
The Sec1/Munc18-1 protein family, which includes significant regulators of the secretory and synaptic vesicle fusion machinery governing hormonal and neuronal transmission, respectively, includes syntaxin-binding protein 1 (STXBP1). Numerous neurological illnesses are linked to STXBP1 pathogenic mutations. Here, we describe the case of a Japanese girl who was born at 40 weeks gestation without experiencing neonatal hypoxia and who had a STXBP1 gene mutation. She experienced generalised seizures and epilepsy at the age of 15 days. She first displayed a series of nodding spasms around the age of 88 days, with the frequency of the seizures rapidly rising. She appeared with developmental regression and the interictal EEG revealed hypsarrhythmia. Genetic testing was carried out at the age of 1.5 years, and mutational analysis identified a STXBP1 gene mutation. She was subsequently determined to have developmental and epileptic encephalopathy, exhibiting the clinical traits of West syndrome brought on by the STXBP1 gene mutation. Her development has remained regressive despite the fact that medication therapy has decreased the frequency of epileptic seizures. It is still unclear how the phenotypic and the type and location of genetic aberration relate to one another. Future research should look at the link between genotype and phenotype as well as the pathophysiology that underlies it in order to clarify the causes of the various phenotype-determining factors.
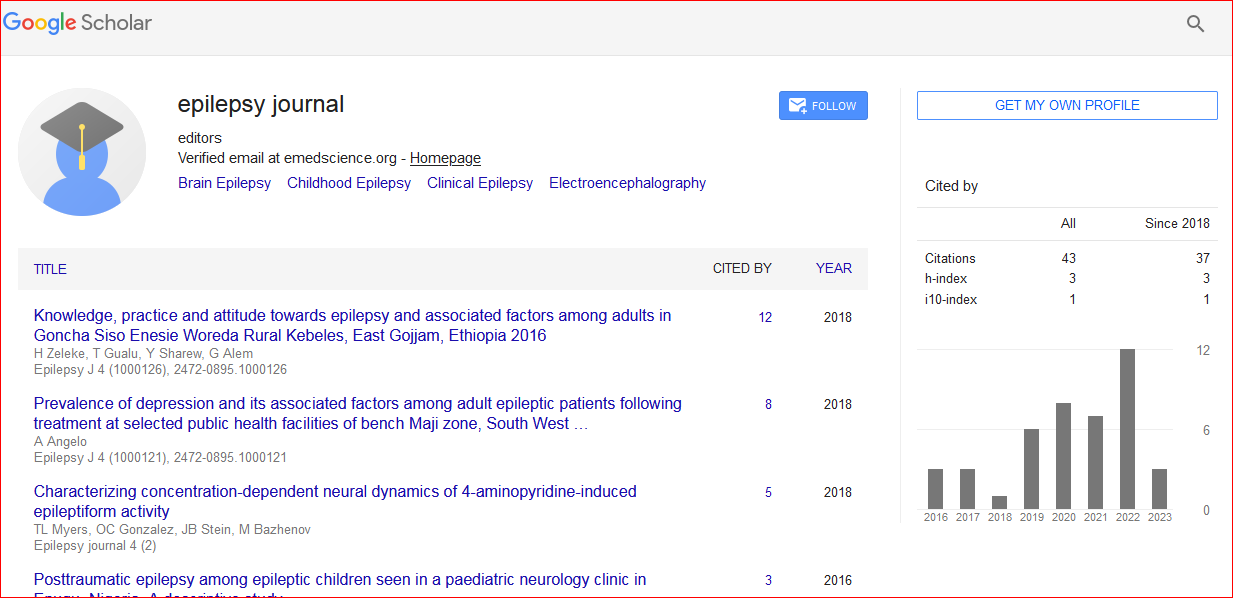
Epilepsy Journal received 41 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 