Extreme cases of intractable childhood epilepsy might culminate in an illness called epileptic encephalopathy. Along with being fatal in certain instances, the illness can result in significant delays in the development of cognitive, sensory, and motor functions. Early infantile SCN8A encephalopathy is associated with missense mutations in SCN8A, which encodes Nav1.6, a key subunit of the voltage-gated sodium channel in neurons and muscles. In this case report, we describe a 5-month-old child who has a new missense mutation associated with SCN8A encephalopathy. The findings of blood and metabolic testing, electroencephalogram (EEG), and brain magnetic resonance imaging (MRI), with the exception of uncontrollable seizures and autistic characteristics, were all normal. Genetic sequencing should be taken into consideration to determine the underlying genetic origins of these mutations since the phenotypes brought on by these mutations cannot be distinguished by any clinical, neuroimaging, or electrophysiological criteria. The administration of oxcarbazepine, as opposed to phenytoin, which is advised as a lastresort treatment for SCN8A encephalopathy, reduced this patient's uncontrollable seizures.
HTML PDFShare this article
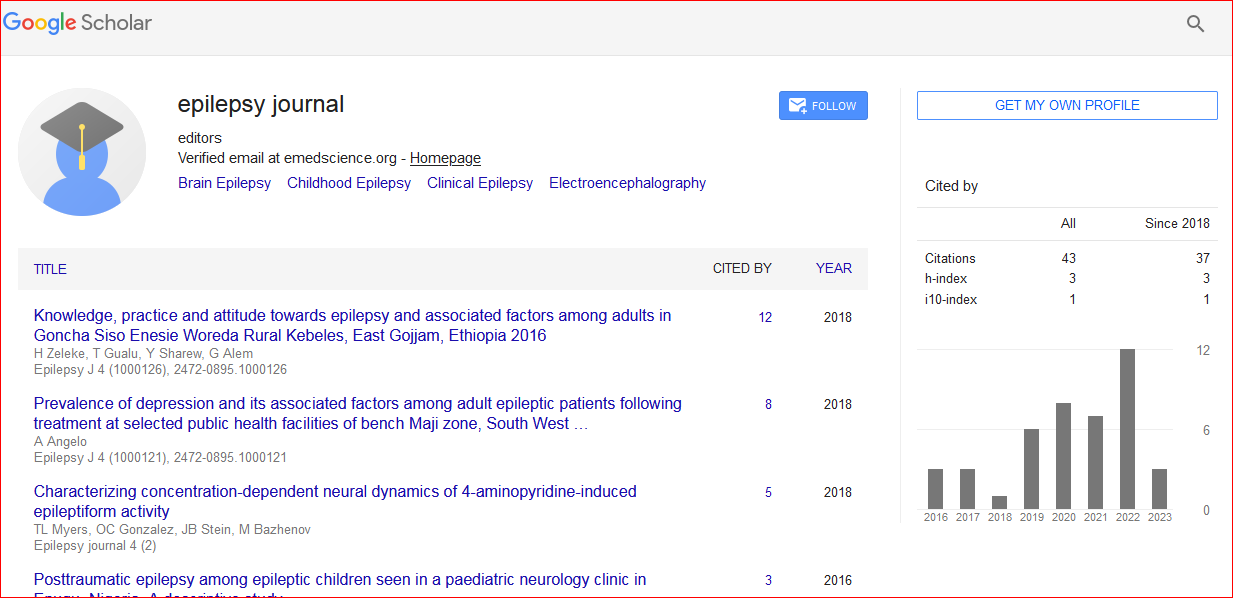
Epilepsy Journal received 41 citations as per Google Scholar report