Gengxin Li
Scientific Tracks Abstracts: J Biomet Biostat
We consider the application of an Empirical Bayes Classification method, originally proposed by Efron, to Genome-wide association study risk prediction using the Genetic Analysis Workshop 17 data. A major advantage of using this method is that the effect size distribution for the set of possible features is empirically estimated and all subsequent parameter estimation and risk prediction is guided by this distribution. Here, we generalize the Efron's method to allow for some of the peculiarities of this data. In particular, we introduce two ways of extending Efron's model: weighted empirical Bayes model and joint covariance model that allow the model to properly incorporate the annotation information of single nucleotide polymorphisms (SNPs). In the course of our analysis, several aspects of the possible simulation model are examined, including the identity of the most important genes, the differing effects of synonymous and non-synonymous single nucleotide polymorphisms (SNPs), and the relative roles of covariates and genes in conferring disease risk. Finally, three methods/models addressed here are compared to each other as well as to other classifiers (random forest and neural network).
Gengxin Li has completed her Ph.D. from Michigan State University and postdoctoral studies from Yale University School of Medicine. She is the Assistant Professor of Mathematics and Statistics Department at Wright State University. She has published several papers in reputed journals.
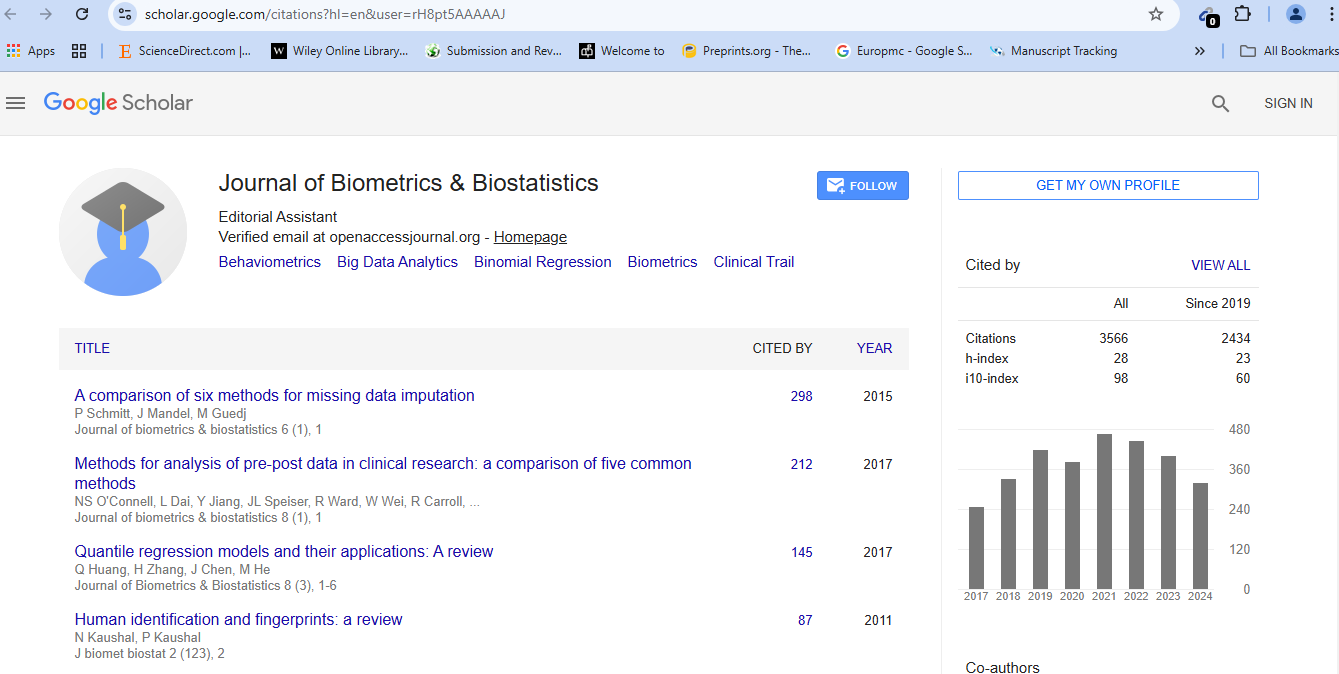
Journal of Biometrics & Biostatistics received 3496 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 