Pervin Unal Civcir
Ankara University, Turkey
Posters & Accepted Abstracts: Chem Sci J
In continuation of our studies of extended heterocyclic systems with potential semi-conductivity and nonlinear optical properties, we have synthesized and studied theoretically some pyrazine derivatives possessing a pyrrole (or thiophene, or furan) ring systems. Extension of the Stetter protocol used in the previous work 1-4 was unsuccessful for the pyrazine analogues because of the poor accessibility of formylpyrazine only gave a complex mixture of products from which the pyrazine could not be isolated.5 We have tried to obtain pyrrolylpyrazine, furylpyridazines, and thienylpyridines from formylpyridazines, via the Paal-Knorr reaction on the intermediary pyrazinyl- 1,4-diketones, using ammonium acetate, polyphosphoric acid or Lawesson's reagent 14, respectively (Scheme 1). The intermediate 1,4-diketone was synthesized via the Stetter reaction in the presence of thiazolium salt as a catalyst. Therefore, we aimed to study theoretically in detail the mechanism of the synthesis for the pyrazine derivatives. In this work, the experimentally suggested mechanisms of Stteter6 and Paal-Knorr7,8 reactions for only pyrrolypyrazine have been modelled with density functional theory (DFT)9, which gives low computational cost and accurate results. Geometries of stationary points along the reaction coordinate have been optimized at BP3LYP10,11 (the hybrid gradient-corrected exchange functional, proposed by Becke, combined with the gradient-corrected correlation functional of Lee, Yang, and Parr) levels of theory using 6-31G* and 6-31G** basis sets12 without any symmetry consideration. Vibrational frequency calculations were used to characterize all stationary points as either minima (no imaginary frequencies) or transition states (with only one imaginary frequency). Vibrational frequencies were also used to evaluate zero point energies and the thermodynamic data. Intrinsic reaction coordinate (IRC) calculations were traced to ensure that the transition states connect the proper minima.13 Mechanisms of the reactions are explained in the potential energy diagrams.
Civcir completed her PhD at the University of East Anglia, UK in 1992 and became a professor in Ankara University in 2009. She is a lecturer in Ankara University, Faculty of Science, Chemistry Department. She works in the field of Organic and Computational Chemistry and has published more than15 papers in reputed journal. She has written a Chemistry book in Turkish. She was involved in translating the five Chemistry books from English to Turkish.
Email:pcivcir@yahoo.com
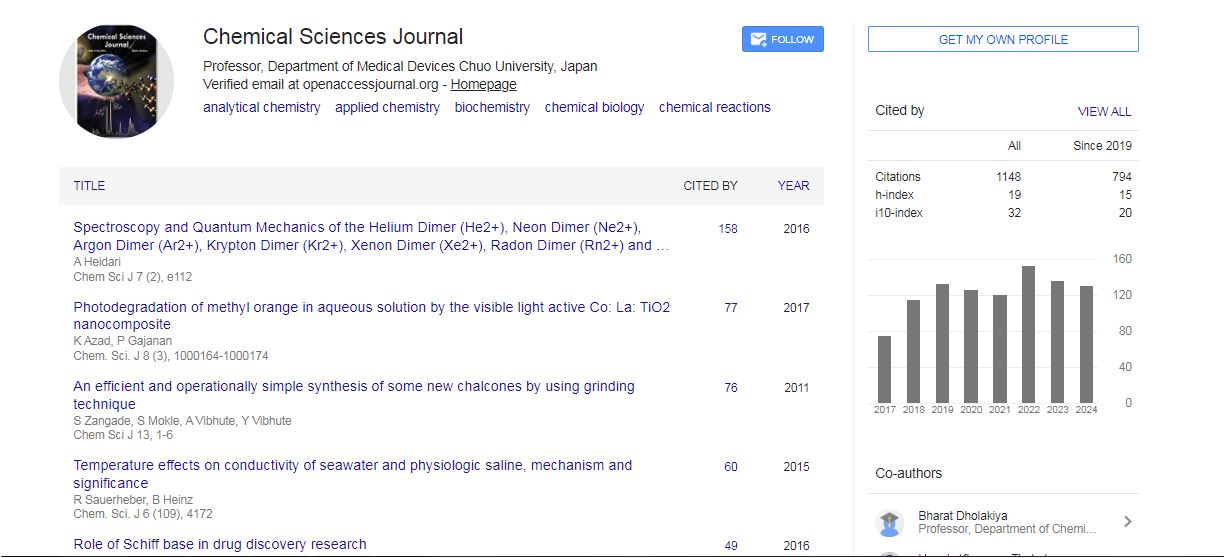
Chemical Sciences Journal received 912 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 