Short Communication - (2025) Volume 9, Issue 3
Received: 01-May-2025, Manuscript No. hps-26-184441;
Editor assigned: 05-May-2025, Pre QC No. P-184441;
Reviewed: 19-May-2025, QC No. Q-184441;
Revised: 22-May-2025, Manuscript No. R-184441;
Published:
29-May-2025
, DOI: 10.37421/2573-4563.2025.9.350
Citation: Silva, Maria P.. ”Pancreatitis: Molecular Pathways, Genetics, and Therapies.” J Hepatol Pancreat Sci 09 (2025):350.
Copyright: © 2025 Silva P. Maria This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Acute pancreatitis (AP) is a rapidly developing inflammatory condition of the pancreas, often initiated by gallstones or alcohol consumption, which can lead to widespread inflammatory responses throughout the body and potentially result in organ failure. This condition necessitates a thorough understanding of its underlying molecular mechanisms to guide effective treatment strategies [1].
Chronic pancreatitis (CP) is characterized by irreversible damage to pancreatic tissue, including the development of fibrosis and the progressive loss of both exocrine and endocrine functions. This chronic state is typically a consequence of repeated episodes of acute injury or is influenced by genetic predispositions, underscoring the long-term impact of pancreatic insults [1].
The molecular underpinnings of pancreatitis involve the premature activation of intracellular digestive enzymes within the pancreatic acinar cells. This auto-digestion process triggers the release of inflammatory mediators, such as cytokines and chemokines, which recruit immune cells to the injured site, thereby amplifying tissue damage [1].
Autophagy, a cellular process for degrading damaged components, and mitochondrial dysfunction have emerged as critical players in both the initiation and progression of pancreatitis. Therapeutic approaches are now increasingly focusing on these cellular pathways to modulate disease activity [1].
Recent research has highlighted the crucial role of acinar cell injury and the premature activation of digestive zymogens as primary events in the onset of acute pancreatitis. The complex interactions between acinar cells, immune cells, and other organs, especially the gut microbiome and the liver, significantly influence the overall severity of the disease [2].
Genetic factors, including mutations in genes such as PRSS1, SPINK1, and CFTR, are increasingly being recognized for their substantial contribution to the development of chronic pancreatitis. This recognition emphasizes the importance of identifying and understanding hereditary forms of the disease for risk assessment and management [2].
Mitochondrial dysfunction is a central pathogenic event observed in both acute and chronic pancreatitis. It contributes to increased oxidative stress, cellular energy depletion, and the amplification of inflammatory responses, creating a vicious cycle of damage [3].
The inflammasome, particularly the NLRP3 inflammasome, plays a pivotal role in orchestrating the inflammatory cascade characteristic of acute pancreatitis. Its activation leads to the release of potent pro-inflammatory cytokines like IL-1β and IL-18, which drive tissue injury and systemic inflammation [4].
Autophagy's role in pancreatitis is multifaceted and context-dependent. While it can serve a protective function by removing damaged organelles and protein aggregates, its dysregulation can paradoxically worsen inflammation and contribute to cell death, making it a complex therapeutic target [5].
The gut microbiome's influence on pancreatitis is a rapidly evolving area of research. An imbalance in the gut's microbial community, known as dysbiosis, can exacerbate systemic inflammation and impact disease severity through various mechanisms, including increased intestinal permeability and immune modulation [6].
Acute pancreatitis (AP) is defined by a rapid onset of pancreatic inflammation, frequently precipitated by gallstones or excessive alcohol intake. This acute inflammatory process can cascade into systemic inflammatory responses, posing a significant risk of multi-organ failure. Understanding the initial cellular events, such as the premature activation of digestive enzymes within acinar cells, is key to comprehending the disease's pathogenesis [1].
Chronic pancreatitis (CP) represents an irreversible condition marked by progressive tissue damage, characterized by fibrosis and the gradual loss of exocrine and endocrine pancreatic functions. This chronic deterioration is often the result of recurrent acute insults or is linked to specific genetic predispositions, highlighting the long-term consequences of pancreatic inflammation [1].
The molecular mechanisms driving pancreatitis involve the intracellular activation of digestive zymogens within acinar cells. This premature activation leads to cellular auto-digestion and the release of inflammatory mediators, including cytokines and chemokines. These signaling molecules attract immune cells, further intensifying the inflammatory process and exacerbating tissue injury [1].
Cellular processes such as autophagy and mitochondrial function play crucial roles in both the initiation and progression of pancreatitis. Dysregulation of autophagy can impair the cell's ability to clear damaged components, while mitochondrial dysfunction leads to energy deficits and oxidative stress, both contributing to disease severity. Therapeutic strategies are increasingly targeting these pathways [1].
Recent advancements have underscored the central role of acinar cell injury and the premature activation of digestive enzymes in initiating acute pancreatitis. Furthermore, the intricate crosstalk between acinar cells, inflammatory cells, and other organs, particularly the gut microbiome and the liver, is critical in determining the severity of the disease [2].
Genetic factors are increasingly recognized as significant contributors to pancreatitis, especially chronic forms. Mutations in specific genes like PRSS1, SPINK1, and CFTR are strongly associated with an increased risk and severity of pancreatitis, emphasizing the importance of hereditary predispositions in disease development [2].
Mitochondrial dysfunction is a key pathogenic event in both acute and chronic pancreatitis. It contributes to oxidative stress, cellular energy depletion, and the amplification of inflammatory responses by releasing damage-associated molecular patterns (DAMPs) that activate immune cells and perpetuate inflammation [3].
The NLRP3 inflammasome is a critical component of the inflammatory cascade in acute pancreatitis. Its activation leads to the processing and release of pro-inflammatory cytokines, such as IL-1β and IL-18, which are instrumental in driving tissue injury and systemic inflammation, making it a prime target for therapeutic intervention [4].
Autophagy exhibits a complex, context-dependent role in pancreatitis. While it can be protective by facilitating the removal of damaged organelles and protein aggregates, its dysregulation can exacerbate inflammation and cell death. Therefore, modulating autophagy pathways requires careful consideration of the specific disease context [5].
The gut microbiome's influence on pancreatitis is a growing area of research. Dysbiosis, an imbalance in gut bacteria, can promote systemic inflammation and worsen disease severity. Mechanisms include increased intestinal permeability and the translocation of bacterial products, leading to altered immune responses [6].
Pancreatitis, encompassing acute and chronic forms, involves complex inflammatory processes within the pancreas. Acute pancreatitis is characterized by rapid inflammation often triggered by gallstones or alcohol, leading to systemic responses and potential organ failure. Chronic pancreatitis involves irreversible tissue damage and functional loss due to repeated insults or genetic factors. Key molecular mechanisms include premature activation of digestive enzymes within acinar cells, leading to auto-digestion and release of inflammatory mediators. Autophagy and mitochondrial dysfunction play critical roles, influencing disease initiation and progression. The inflammasome, particularly NLRP3, is central to the inflammatory cascade in acute pancreatitis, releasing pro-inflammatory cytokines. Genetic predispositions through mutations in genes like PRSS1, SPINK1, and CFTR are significant contributors to chronic pancreatitis. The gut microbiome also plays a role, with dysbiosis exacerbating inflammation. Therapeutic strategies are increasingly focused on targeting these molecular pathways, including autophagy, mitochondrial health, and anti-inflammatory approaches, with ongoing clinical trials evaluating their efficacy.
Pancreatitis, encompassing acute and chronic forms, involves complex inflammatory processes within the pancreas. Acute pancreatitis is characterized by rapid inflammation often triggered by gallstones or alcohol, leading to systemic responses and potential organ failure. Chronic pancreatitis involves irreversible tissue damage and functional loss due to repeated insults or genetic factors. Key molecular mechanisms include premature activation of digestive enzymes within acinar cells, leading to auto-digestion and release of inflammatory mediators. Autophagy and mitochondrial dysfunction play critical roles, influencing disease initiation and progression. The inflammasome, particularly NLRP3, is central to the inflammatory cascade in acute pancreatitis, releasing pro-inflammatory cytokines. Genetic predispositions through mutations in genes like PRSS1, SPINK1, and CFTR are significant contributors to chronic pancreatitis. The gut microbiome also plays a role, with dysbiosis exacerbating inflammation. Therapeutic strategies are increasingly focused on targeting these molecular pathways, including autophagy, mitochondrial health, and anti-inflammatory approaches, with ongoing clinical trials evaluating their efficacy.
None
None
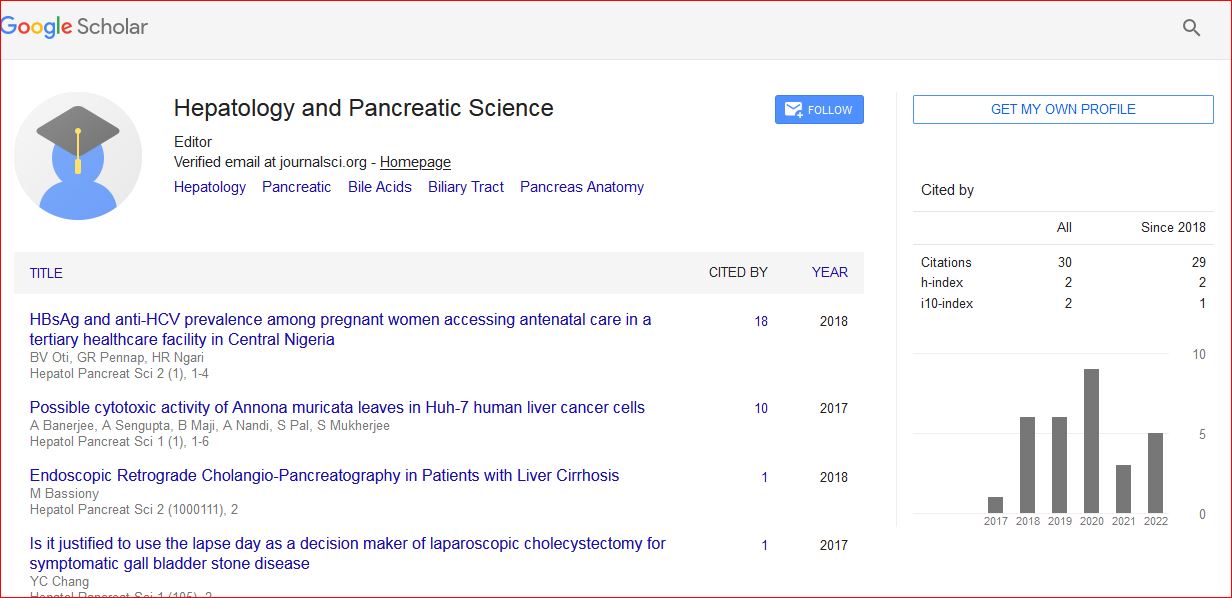
Hepatology and Pancreatic Science received 34 citations as per Google Scholar report