Brief Report - (2025) Volume 11, Issue 6
Received: 01-Nov-2025, Manuscript No. ldt-25-178459;
Editor assigned: 03-Nov-2025, Pre QC No. P-178459;
Reviewed: 17-Nov-2025, QC No. Q-178459;
Revised: 24-Nov-2025, Manuscript No. R-178459;
Published:
29-Nov-2025
, DOI: 10.37421/2472-1018.2025.11.333
Citation: Romano, Luca. ”Endothelial Dysfunction: Key to Lung Disease Pathogenesis.” J Lung Dis Treat 11 (2025):333.
Copyright: © 2025 Romano L. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Endothelial dysfunction represents a pivotal early stage in the pathogenesis of numerous lung ailments. This compromised state of the endothelium underlies a cascade of detrimental vascular events, including aberrant vascular tone, heightened permeability, pervasive inflammation, and the promotion of pro-thrombotic conditions within the pulmonary vasculature. Consequently, this dysfunction significantly contributes to the inception and advancement of serious conditions such as pulmonary arterial hypertension, chronic obstructive pulmonary disease (COPD), asthma, and acute respiratory distress syndrome (ARDS), by disrupting the finely tuned balance governing vasodilation and vasoconstriction, immune cell trafficking, and fluid equilibrium within the lungs [1].
Within the context of COPD, the impact of endothelial dysfunction is not confined to the pulmonary arteries but extends to the systemic circulation. This systemic endothelial compromise is implicated in the development of significant comorbidities, including cardiovascular disease and skeletal muscle atrophy, thereby adversely affecting the overall health status and prognostic outlook of patients diagnosed with COPD. The exploration of therapeutic strategies aimed at modulating endothelial function thus emerges as a promising avenue for mitigating these widespread systemic manifestations [2].
The intricate inflammatory cascade characteristic of asthma involves a substantial degree of endothelial activation and subsequent dysfunction. An increase in vascular permeability is a key consequence, leading to the accumulation of fluid within the airways and the exacerbation of airway edema, a hallmark feature of asthma exacerbations. Endothelial cells actively participate in the recruitment of inflammatory cells to the airway mucosa, thereby perpetuating the chronic inflammatory process that defines the disease. Consequently, therapeutic approaches focused on restoring endothelial function and curtailing vascular leakage are actively under investigation [3].
Acute Respiratory Distress Syndrome (ARDS) is fundamentally characterized by widespread alveolar damage and intense inflammation, during which the integrity of the endothelial barrier in the pulmonary capillaries is severely compromised. Endothelial cells lining these capillaries become activated, leading to a marked increase in permeability, subsequent plasma extravasation into the alveolar spaces, and a critical impairment of gas exchange. A thorough understanding of the underlying mechanisms driving this endothelial barrier dysfunction is therefore paramount for the development of effective therapeutic interventions for ARDS [4].
Oxidative stress stands out as a principal instigator of endothelial dysfunction across a broad spectrum of lung diseases. This detrimental process leads to the uncoupling of nitric oxide synthase, a critical enzyme involved in vascular homeostasis, resulting in a reduced bioavailability of nitric oxide (NO). Simultaneously, it augments the production of reactive oxygen species, collectively contributing to significant vascular impairment. Therefore, therapeutic strategies designed to mitigate oxidative stress hold substantial promise for conferring protection against lung injury and impeding disease progression [5].
Inflammation is intrinsically interwoven with the development of endothelial dysfunction in various pulmonary pathologies. Inflammatory mediators, notably cytokines, exert their influence by activating endothelial cells, thereby promoting the expression of adhesion molecules essential for leukocyte binding and transmigration. This process, coupled with the facilitation of microthrombosis, establishes an inflammatory feedback loop that exacerbates lung injury and amplifies disease severity [6].
A notable characteristic of endothelial dysfunction in lung diseases is the impairment of nitric oxide (NO) signaling. This deficit can arise from either reduced NO production or accelerated NO degradation, both of which compromise NO's crucial roles in maintaining vasodilation, inhibiting platelet aggregation, and exerting anti-inflammatory effects. Consequently, deficiencies in NO bioactivity contribute directly to pathological pulmonary vasoconstriction and adverse vascular remodeling [7].
Microvascular dysfunction plays a particularly significant role in the pathogenesis of interstitial lung diseases. The reduction in capillary density and the consequent impairment of blood flow within the lung interstitium can precipitate hypoxia and induce tissue damage, thereby contributing to disease progression and the development of fibrosis. In this context, endothelial cells are indispensable for preserving the structural integrity and functional capacity of these vital microvessels [8].
Therapeutic interventions specifically designed to target endothelial dysfunction have demonstrated considerable promise in improving clinical outcomes for certain lung diseases, most notably pulmonary arterial hypertension. Examples include the judicious use of statins, phosphodiesterase-5 inhibitors, and prostacyclin analogues. However, further extensive research is warranted to fully elucidate and optimize their broader therapeutic applications across the diverse landscape of pulmonary conditions [9].
The endothelium's role in orchestrating and maintaining lung homeostasis cannot be overstated. Any disruption to its crucial barrier function, alterations in its vasoreactivity, or the establishment of pro-inflammatory signaling pathways within this layer significantly contributes to the complex pathophysiology of a wide array of lung diseases. Consequently, the endothelium represents a vital and highly promising area for ongoing scientific investigation and the development of novel therapeutic strategies [10].
Endothelial dysfunction serves as a critical early event in the pathological development of numerous lung diseases. This dysfunction underlies a complex interplay of vascular abnormalities, including impaired vascular tone, increased permeability, significant inflammation, and the promotion of pro-thrombotic states within the pulmonary vasculature. These cellular and molecular changes contribute substantially to the initiation and progression of conditions such as pulmonary arterial hypertension, COPD, asthma, and ARDS, by disrupting the normal physiological balance of vasodilation and vasoconstriction, regulating immune cell trafficking, and maintaining fluid balance within the delicate lung environment [1].
In the context of Chronic Obstructive Pulmonary Disease (COPD), the detrimental effects of endothelial dysfunction transcend the pulmonary arteries and extend into the systemic circulation. This widespread systemic endothelial impairment is a significant contributor to the development of severe comorbidities, including cardiovascular disease and skeletal muscle atrophy. These systemic manifestations profoundly impact the overall health and long-term prognosis of individuals suffering from COPD. Therefore, targeting endothelial function emerges as a potentially groundbreaking therapeutic strategy to ameliorate these pervasive systemic consequences [2].
The inflammatory processes central to the pathogenesis of asthma are intrinsically linked with significant endothelial activation and subsequent dysfunction. A hallmark of this is the increase in vascular permeability, which directly leads to airway edema, a key clinical manifestation during asthma exacerbations. Furthermore, endothelial cells play a crucial role in orchestrating the inflammatory response by facilitating the recruitment of circulating inflammatory cells to the airways, thereby perpetuating the chronic disease state. Consequently, therapeutic strategies aimed at restoring endothelial function and reducing excessive vascular leakage are actively being explored as potential treatments [3].
Acute Respiratory Distress Syndrome (ARDS) is characterized by diffuse alveolar damage and a robust inflammatory response, wherein the integrity of the endothelial barrier in the pulmonary capillaries is severely compromised. Endothelial cells lining the lung capillaries become activated, leading to a pronounced increase in vascular permeability, resulting in the leakage of plasma into the alveoli and critically impairing the efficiency of gas exchange. Therefore, a comprehensive understanding of the molecular and cellular mechanisms that drive endothelial barrier dysfunction is essential for the successful development of effective therapeutic interventions for ARDS [4].
Oxidative stress represents a primary driving force behind endothelial dysfunction observed in a diverse range of lung diseases. Its detrimental effects include the uncoupling of nitric oxide synthase, leading to a significant reduction in the bioavailability of nitric oxide, and an amplified production of reactive oxygen species. These processes collectively result in substantial vascular impairment. Accordingly, therapeutic interventions focused on reducing oxidative stress hold considerable promise for protecting the lungs against injury and mitigating disease progression [5].
Inflammation is fundamentally intertwined with endothelial dysfunction in the context of pulmonary pathologies. Inflammatory mediators, such as cytokines, potently activate endothelial cells, thereby enhancing the expression of adhesion molecules that promote leukocyte adhesion and transmigration across the vascular wall. This inflammatory process, often accompanied by the development of microthrombosis, forms a vicious cycle that exacerbates lung injury and increases disease severity [6].
An impaired nitric oxide (NO) signaling pathway, resulting from either diminished NO production or increased NO degradation, is a defining feature of endothelial dysfunction in many lung diseases. NO plays indispensable roles in maintaining vascular homeostasis by promoting vasodilation, inhibiting platelet aggregation, and exerting anti-inflammatory effects. Deficiencies in NO bioactivity directly contribute to pathological pulmonary vasoconstriction and adverse vascular remodeling, further compromising lung function [7].
Microvascular dysfunction significantly contributes to the pathophysiology of interstitial lung diseases. The observed reduction in capillary density and the consequent impairment of blood flow within the lung interstitium can lead to tissue hypoxia and ongoing damage, thereby exacerbating disease progression and promoting the development of fibrosis. In this critical microenvironment, endothelial cells are central to maintaining the structural integrity and functional capacity of these vital lung microvessels [8].
Therapeutic strategies designed to address endothelial dysfunction have shown notable promise in improving patient outcomes in specific lung diseases, particularly pulmonary arterial hypertension. These interventions include the use of established medications such as statins, phosphodiesterase-5 inhibitors, and prostacyclin analogues. Nevertheless, further rigorous research is imperative to explore and optimize their broader applicability across the spectrum of other pulmonary conditions [9].
The endothelial lining of the lungs plays a paramount role in maintaining overall lung homeostasis. Any disruption to its critical barrier function, coupled with aberrant vasoreactivity and the establishment of pro-inflammatory signaling pathways, significantly contributes to the complex pathophysiology of a wide variety of lung diseases. Consequently, the endothelium represents a vital and strategically important area for continued scientific investigation and the development of innovative therapeutic interventions [10].
Endothelial dysfunction is a critical early event in many lung diseases, contributing to impaired vascular tone, increased permeability, inflammation, and pro-thrombotic states. This dysfunction is implicated in pulmonary arterial hypertension, COPD, asthma, and ARDS. In COPD, it extends to systemic circulation, affecting comorbidities. Asthma involves endothelial activation leading to airway edema and inflammation. ARDS is characterized by compromised endothelial barrier integrity and plasma leakage. Oxidative stress and inflammation are major drivers of endothelial dysfunction in lung diseases. Impaired nitric oxide signaling is a hallmark, leading to vasoconstriction and remodeling. Microvascular dysfunction is significant in interstitial lung diseases, causing hypoxia and fibrosis. Therapeutic interventions targeting endothelial dysfunction, like statins and NO-enhancing drugs, show promise, especially in pulmonary arterial hypertension. The endothelium's role in lung homeostasis makes it a vital area for research and therapeutic development.
None
None
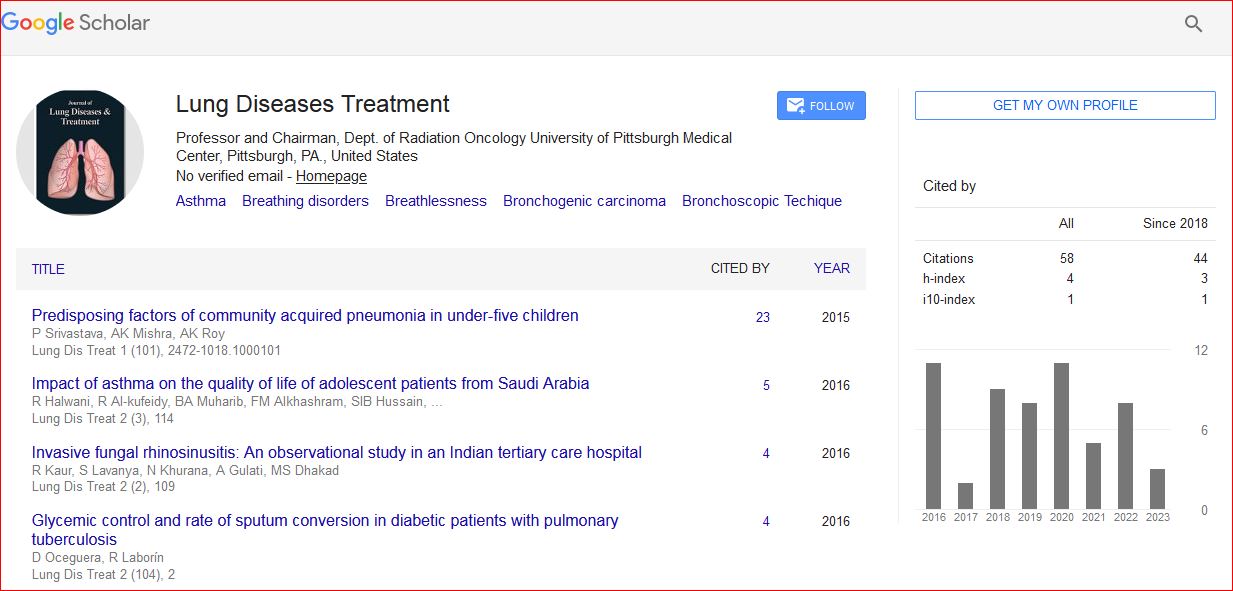
Journal of Lung Diseases & Treatment received 247 citations as per Google Scholar report