Bernard Rosner, Wei Wang, Heather Eliassen and Eileen Hibert
DOI: 10.4172/2155-6180.1000226
There already exist methods for comparing dependent Pearson correlation coefficients. However, each of the variables (X, Y) has associated random error; and a related question is after correcting for random error, which variable correlates most highly with the outcome variable Z. In this paper, we present methods for comparing dependent deattenuated correlation coefficients. This is a generalization of previous work for obtaining confidence limits for a single deattenuated correlation coefficient. In addition, we extend this work to the comparison of dependent Spearman correlation coefficients. The methods are illustrated with two examples. The first example concerns the comparison of nephrotoxicity of phenacetin and aspirin intake as measured by repeat biomarkers obtained from the same subjects. The second example is a comparison of the validity of different storage conditions for measuring HbA1c from dried blood specimens as compared to the gold standard of immediate processing. Results from using these methods indicate that phenacetin intake is more highly correlated with serum creatinine levels than aspirin intake and that short-term storage is preferable to long-term storage for assessment of HbA1c levels. We have available SAS software for comparing dependent deattenuated Pearson correlation and dependent Spearman correlations with and without deattentuation.
Liu J, Wang F, Gao X, Zhang H, Wan X and Can Yang
DOI: 10.4172/2155-6180.1000228
Over one thousand genome-wide association studies (GWAS) have been conducted in the past decade. Increasing biological evidence suggests the polygenic genetic architecture of complex traits: a complex trait is affected by many risk variants with small or moderate effects jointly. Meanwhile, recent progress in GWAS suggests that complex human traits may share common genetic bases, which is known as “pleiotropy”. To further improve statistical power of detecting risk genetic variants in GWAS, we propose a penalized regression method to analyze the GWAS dataset of primary interest by incorporating information from other related GWAS. The proposed method does not require the individual-level of genotype and phenotype data from other related GWAS, making it useful when only summary statistics are available. The key idea of the proposed approach is that related traits may share common genetic basis. Specifically, we propose a linear model for integrative analysis of multiple GWAS, in which risk genetic variants can be detected via identification of nonzero coefficients. Due to the pleiotropy effect, there exist genetic variants affecting multiple traits, which correspond to a consistent nonzero pattern of coefficients across multiple GWAS. To achieve this, we use a group Lasso penalty to identify this nonzero pattern in our model, and then develop an efficient algorithm based on the proximal gradient method. Simulation studies showed that the proposed approach had satisfactory performance. We applied the proposed method to analyze a body mass index (BMI) GWAS dataset from a European American (EA) population and achieved improvement over single GWAS analysis.
DOI: 10.4172/2155-6180.1000229
Phase II cancer clinical trials are conducted for initial evaluation of the therapeutic efficacy of a new treatment regimen, and two-stage designs often are implemented in such trials. Typically, designs of phase II trials not only satisfy predefined significance and power requirements but also have some desirable features such as minimizing the total sample size, minimizing the average sample size under the null hypothesis, etc. A frequent issue is that the attained sample sizes differ from the planned sample sizes. We propose alternative designs adjusted to the attained sample sizes when they are different than the planned sample sizes. We present extensive examples and compare the proposed designs to that of Green and Dahlberg. We apply the proposed designs to a phase II trial in non-Hodgkin’s lymphoma patients.
Nevis IF, Sikich N, Ye C and Kabali C
DOI: 10.4172/2155-6180.1000230
Background: Systematic reviews (SRs) remain the core of evidence based medicine. Routinely, two reviewers are required to screen titles and abstracts in SRs. Nonetheless, many organizations may use a single reviewer due to restricted time and resources. In situations where there is only a single reviewer, we propose a sampling method and assessed its performance in a simulation study. Methods: We described the sampling process guided by a set of instructions. For validation, we generated 20,000 citations from a skewed normal distribution and assigned a score of raters’ agreement. From these, we randomly selected a fixed number of citations, whose probability of selection was determined by a uniform distribution, and repeated the iteration 1000 times. In each iteration set, the sample size was fixed at 50, 100, and 200. Results: We evaluated the sampling performance and proposed the appropriate sample size formula. Of the 20,000 citations, 86.7% fell into the category of “both reviewers have an agreement”. The sampling performance was optimal. On average, the percent of agreement for samples of size 50, 100, and 200 were 86.7% (95% CI 76% to 96%), 86.7% (95% CI 79% to 93%), and 86.8% (95% CI 81.5% to 91.5%) respectively. When comparing the performance of sample size formula with simulations, we obtained identical results. Conclusions: We propose a reliable and valid sampling methodology for screening titles and abstracts This method may be used in resource constrained environments conducting SRs.
Bohning D and Viwatwongkasem C
DOI: 10.4172/2155-6180.1000231
This note shows that the concept of an offset, frequently introduced in Poisson regression models to cope with ratetype data, can be simply treated with a regular Poisson regression model. Hence Poisson regression models requiring an offset can be fitted with ordinary Poisson regression models. Some illustrations are provided and it is discussed how this result came about.
DOI: 10.4172/2155-6180.1000232
The Nelson-Aalen estimator provides the basis for the ubiquitous Kaplan-Meier estimator, and therefore is an essential tool for nonparametric survival analysis. This article reviews martingale theory and its role in demonstrating that the Nelson-Aalen estimator is uniformly consistent for estimating the cumulative hazard function for right-censored continuous time-to-failure data.
Shoukri MM, Collison K and Al-Mohanna F
DOI: 10.4172/2155-6180.1000233
Background: The metabolic syndrome is intimately linked hypertension, impaired glucose tolerance, dyslipidemia, and abdominal obesity and is associated with an increased risk of total and cardiovascular mortality in adults. Genetics as well as environmental influences have been implicated in obesity and several cardiovascular risk factors. Because Family is one of the most important factors affecting metabolic risk factors, studying co-aggregation of the components of the syndrome among family members, and in particular spousal pairs, is of interest to genetic epidemiologists and community health researchers. Methods: Based on the clinical definition of the syndrome, we introduce three statistical models to estimate the prime parameter of interest which measure the degree of clustering of the disease among spousal pairs. Since the focus in this paper is on the methodological approach to estimate the between pairs clustering parameters, we shall use Monte-Carlo simulated data for demonstration purposes, with values of the input parameters for each component taken from a well-known Korean study. We develop two models, the Bivariate Truncated Poisson Model (BTPM), which models the counts, and the Bivariate Dirichlet Multinomial Model (BDMM), which models the frequency of counts, and discuss the relative merits of each model. The two models are qualitatively different but quantitatively interrelated. Since the clinical definition of the metabolic syndrome requires that at least three of its components, co-exist within a subject, we show that adhering to this definition requires certain specifications that should be satisfied in any of the adopted models. We estimated the clustering parameters under the specified models. A comparison between the models was based on the internal consistency of each model. What we mean by that is the degree of closeness of the estimated distribution to the observed data. The BDMM fitted the data much closer than the BTPM. Interpretation: In a sample of randomly selected spousal pairs; and according to the clinical definition, the number of components of the metabolic syndrome can in an individual be 0, 1, 2, 3, 4, or 5. Estimation of the clustering parameter of the counts is equivalent to the estimation of the intraclass correlation coefficient (ICCC) between pairs. Assessing the goodness of fit of the proposed models, it is more statistically sound to estimate the degree of clustering of the components of the syndrome in spousal pairs under the BDMM.
Szczesniak RD, Li D and Raouf SA
DOI: 10.4172/2155-6180.1000234
The potential to characterize nonlinear progression over time is now possible in many health conditions due to advancements in medical monitoring and more frequent data collection. It is often of interest to investigate differences between experimental groups in a study or identify the onset of rapid changes in the response of interest using medical monitoring data; however, analytic challenges emerge. We review semiparametric mixed-modeling extensions that accommodate medical monitoring data. Throughout the review, we illustrate these extensions to the semiparametric mixed-model framework with an application to prospective clinical data obtained from 24-hour ambulatory blood pressure monitoring, where it is of interest to compare blood pressure patterns from children with obstructive sleep apnea to those arising from healthy controls.
Liu X, Freed MC and McCutchan PK
DOI: 10.4172/2155-6180.1000235
Researchers often encounter discrete response data in longitudinal analysis. Generalized linear mixed models are generally applied to account for potential lack of independence inherent in longitudinally data. When parameter estimates are used to describe longitudinal processes, random effects, both between and within subjects, need to be retransformed in nonlinear predictions on the response data; otherwise, serious retransformation bias can arise to an unanticipated extent. This study attempts to go beyond existing work by developing a retransformation method deriving statistically robust longitudinal trajectory of nonlinear predictions. Variances of population-averaged nonlinear predictions are approximated by the delta method. The empirical illustration uses longitudinal data from the Asset and Health Dynamics among the Oldest Old study. Our analysis compares three sets of nonlinear predictions of death rate at six time points, from the retransformation method, the best linear unbiased predictor, and the fixed-effects approach, respectively. The results demonstrate that failure to retransform the random components in generalized linear mixed models results in severely biased nonlinear predictions, as well as much reduced standard error approximates.
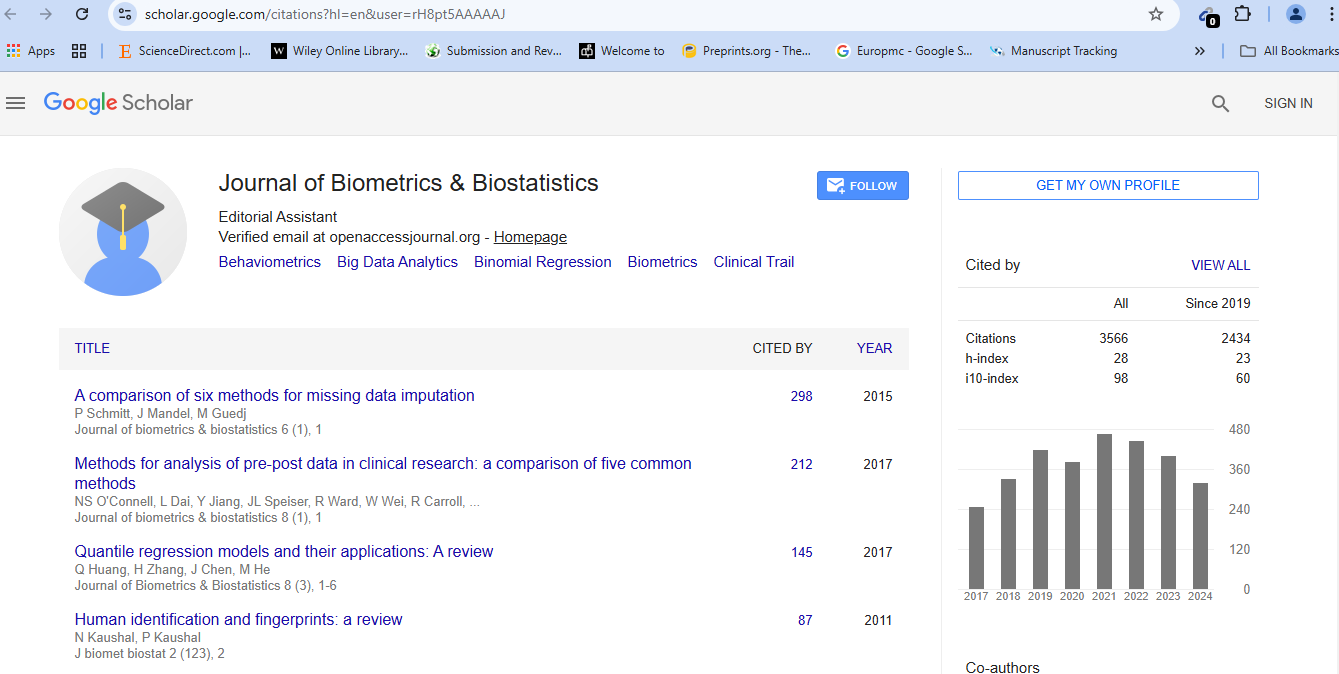
Journal of Biometrics & Biostatistics received 3496 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 