Sudheendra Kulkarni
Syngene Clinical Development, India
Scientific Tracks Abstracts: J Bioanal Biomed
Clinical Research Organizations (CRO) supporting the biopharmaceutical and pharmaceutical industry has seen a paradigm shift in how regulatory inspections are conducted. The current expectation is that the CRO has to demonstrate quality and data integrity (DI) in order to gain the trust of the regulatory investigators. Adherence to DI standards has become one of the key areas of focus during regulatory inspections. The systems used in the research industry have become more complex in terms of metadata that is being generated across the companies. Metadata that is created in a clinical trial is quite voluminous and the inspectors are aware of the possible DI issues that can arise from time-to-time. The functions such as user access, delete features data backup policies, audit trail, role of the system administrator, etc., have become the focus of attention during inspections. ALCOA principles are applicable to both paper and electronic data, thereby to ensure that the DI standards as part of good documentation practices (GDP) are followed uniformly by all personnel who generate clinical trial data. To ensure that the DI standards are maintained across the life cycle of the clinical trial, quality systems should be integrated and omnipresent in the operation to ensure the protocol, regulations and procedures are followed. Adherence to these standards will ensure that the clinical trial data generated will meet the applicable regulatory requirements. The current climate is such that, the DI requirements shall be more demanding and a primary focal point in clinical research. The focus of this presentation will highlight how we prepared the teams on the current global requirements to ensure a successful regulatory inspection.
Sudheendra K has his expertise in managing the GCP quality management systems at Syngene Clinical Development in the conduct of Phase I, bioequivalence, bioavailability studies. He has hosted many international regulatory inspections such as US FDA, EMA, ANVISA, Thailand GLP at Syngene Clinical Development. He is a trained Biochemist and has been associated with GCP QMS management for the last 14+ years. He has also audited many vendors and service providers who provide support for the contract research organizations for the conduct of early phase studies. He has also audited many complex clinical trials, Phase II and Phase III trials that have been conducted at different hospital sites.
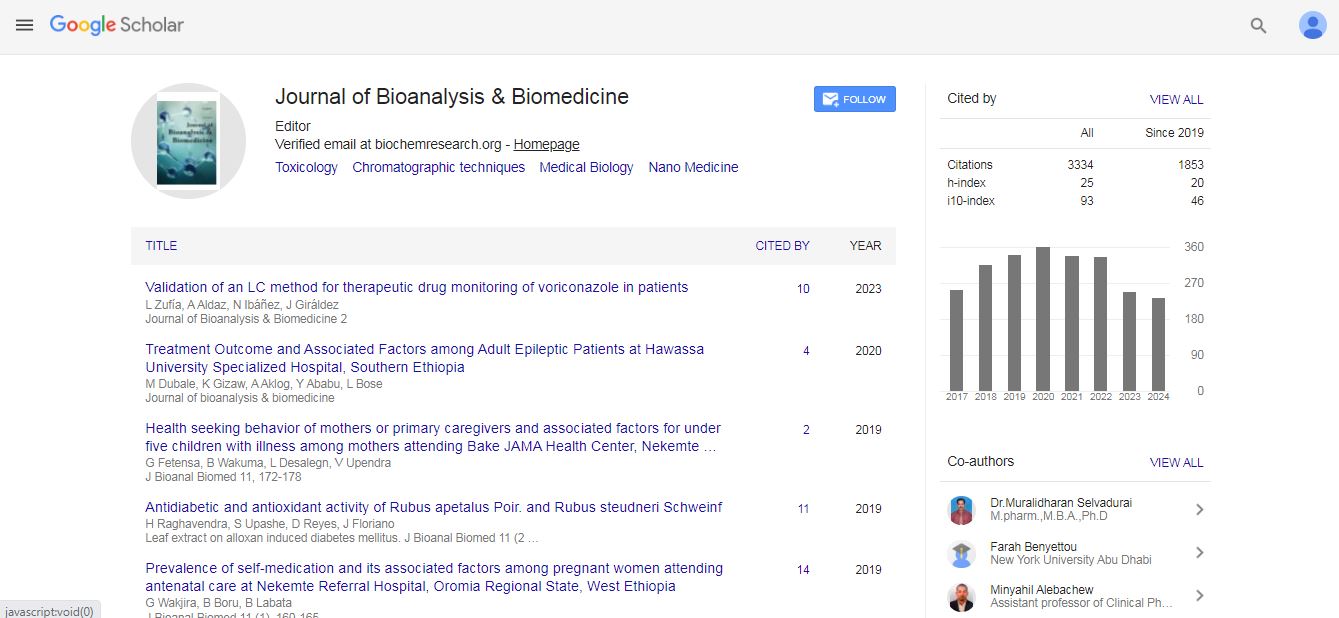
Journal of Bioanalysis & Biomedicine received 3099 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 