J Vijay Venkatraman
Scientific Tracks Abstracts: J Bioanal Biomed
Biosimilars/Biologics are given various names in different countries: Biosimilars in the EU and Australia, Follow-on Biopharmaceuticals in the USA and Japan, Subsequent Entry Biologics in Canada and are called Similar Biotherapeutic Products according to the WHO. A Biosimilar is defined by the European Medicines Agency as a biological medicine that is developed to be similar to an existing medicine (a reference medicine). The first biopharmaceutical products were launched in the 1980s and adopted a manufacturing method of recombinant technologies. These products are now on their way to patent expiration and thus arises the need for the production of biosimilars which are highly similar to the small molecule products derived from recombinant technology and posses higher molecular complexity. The need for pharmacovigilance for such products emerges as these biosimilars require more rigorous assessment when compared to traditional generic medicines. There are various challenges faced by the innovators and the manufacturers of biosimilars to warrant patient safety and to maintain consistency in their production. Since these products may differ in the production process from batch to batch which may hamper purity of the final product, a tailored pharmacovigilance plan must be implemented. The EMA (European Medicines Agency) has taken the precedence in issuing guidelines as precautions, most of which are still under review. The guidelines address pre-clinical and clinical testing prior to market authorization and also postmarketing pharmacovigilance plans are expected to be included. A risk management plan stating the safety data of the product must be submitted by the manufacturers and approved by the regulatory authorities in order to conduct trials and obtain authorization for the particular product. The RMP must be submitted as a part of the marketing application for all new chemical and biological drugs, inclusive of biosimilars. Immunogenicity and lack of efficacy as a safety concern should be included in the RMP and the requirement for additional pharmacovigilance activities should be gauged. When biosimilars are approved in the EU they are considered ?comparables? to the reference product, but are not ensured to bear therapeutic equivalence. In the EU, a total of 14 biosimilars have been approved by the EMA. Adverse event reporting with regard to biosimilars requires the member states to take all measures to identify any biological product dispensed, which is the subject of an adverse reaction report. The traceability is improved by commercializing biologics with a brand name or the international nonproprietary name plus the manufacturer?s name. In accordance with the FDA, biosimilars are products highly similar to a US-licensed reference biological product not withstanding minor differences in clinically inactive components and in which there are no differences between the biological product and the reference product in terms of safety, purity and potency. Till date, the FDA has not approved a biological product as a biosimilar or interchangeable. The personnel at the USFDA are currently working on the review and licensure process for biosimilars. In conclusion, it can be summarized that pharmacovigilance, as a part of risk management programmes, will need to comprehensively include regular monitoring for the consistency, safety, traceability and the efficacy of the drug. Further, postmarketing surveillance in case of biosimilars is critical due to the limited spontaneous information available with regard to suspected adverse drug reactions associated with their usage.
J Vijay Venkatraman is a Diabetologist and Drug Safety Physician with an overall experience of 13 years, of which the last 7 years have been in the Pharmacovigilance industry. He holds a MBA degree in Services Management. In March 2012, Vijay founded Oviya MedSafe Pvt Ltd, a Pharmacovigilance Consulting & Services Company based out of Coimbatore in India, which has been catering to the international as well as the Indian pharmaceutical industry since then. He is an Affiliate Member of the Faculty of Pharmaceutical Medicine of the Royal Colleges of Physicians of the United Kingdom and an Associate Member of the Pharmaceutical Information & Pharmacovigilance Association, United Kingdom. He is an Executive Committee Member of the Indian Society for Clinical Research for 2013-15. He is also the Secretary Elect of the Indian Medical Association ? Coimbatore Branch for 2015.
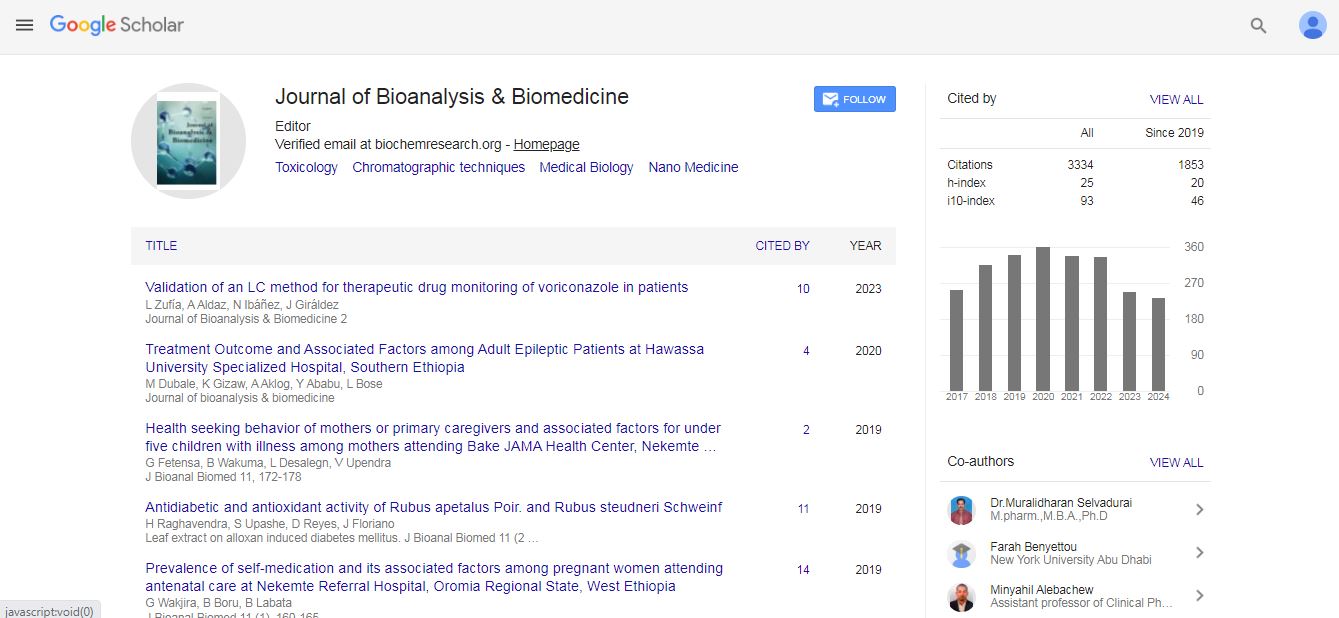
Journal of Bioanalysis & Biomedicine received 3099 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 