Wolfgang A Rehmann
Taylor Wessing, Germany
Posters-Accepted Abstracts: J Bioanal Biomed
In Europe, biosimilars are governed by the statutory provisions applicable for generic medicines. The legal requirements and the procedures for making an application for a marketing authorization are set out in Directive 2016/83/EC and in Regulation (EC) No 726/2004. There are a number of possible approaches to getting approval for a bio-similar product in Europe, in particular to either submit a full dossier of pre-clinical and clinical data as would be required for any new medicinal product or to apply under the abridged procedure. An applicant for approval of a bio-similar product under the abridged procedure is required to demonstrate that its product is similar to a previously authorized product. The EMA is responsible for assessing applications from companies to market biological medicines for use in the European Union, including bio-similar medicines in case the centralized procedure is applicable. The EMA has published guidelines that govern how biosimilars are to be assessed. There are three guidelines that apply to all biosimilars: an overarching guideline which was first adopted in September 2005 and was amended in 2014, coming into effect on 30 April 2015; and two further guidelines which were both adopted by the EMA on 22 February 2006, one relating to non-clinical and clinical issues and one to quality issues. The guideline on quality issues was also amended in 2014. The presentation will give an overview to the audience about how the legal framework in the EU works and will give an update on most recent legal development within the EU in this regard.
Email: W.Rehmann@taylorwessing.com
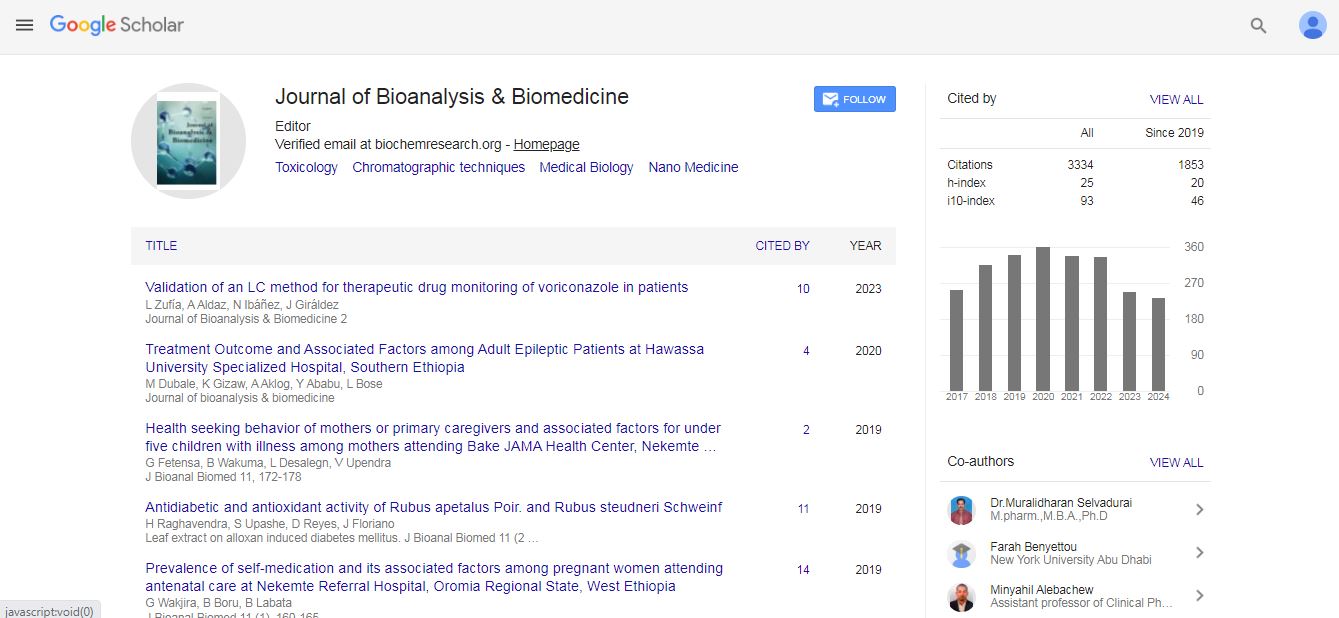
Journal of Bioanalysis & Biomedicine received 3099 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 