Commentary - (2025) Volume 11, Issue 2
Received: 01-Mar-2025, Manuscript No. ldt-25-178407;
Editor assigned: 03-Mar-2025, Pre QC No. P-178407;
Reviewed: 17-Mar-2025, QC No. Q-178407;
Revised: 24-Mar-2025, Manuscript No. R-178407;
Published:
31-Mar-2025
, DOI: 10.37421/2472-1018.2025.11.294
Citation: Zhang, Wei. ”Pulmonary Fibrosis: Molecular Pathways and Therapeutic Strategies.” J Lung Dis Treat 11 (2025):294.
Copyright: © 2025 Zhang W. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Pulmonary fibrosis is a complex chronic lung disease characterized by the progressive scarring of lung tissue, significantly impairing respiratory function. The intricate molecular mechanisms underlying its pathogenesis are multifaceted and involve a network of signaling pathways and cellular interactions that culminate in excessive extracellular matrix deposition [1].
Among the key molecular players, the Transforming Growth Factor-beta (TGF-β) signaling cascade stands out as a central driver of fibrotic processes. It orchestrates a series of events, including fibroblast proliferation and the transformation of quiescent fibroblasts into highly contractile myofibroblasts, which are the primary collagen-producing cells [2].
Inflammatory responses also play a crucial role in initiating and perpetuating lung damage and fibrosis. Various cytokines, such as Interleukin-13 (IL-13) and Tumor Necrosis Factor-alpha (TNF-α), are released in the lung microenvironment, promoting chronic inflammation and contributing to the fibrotic cascade [3].
The abnormal activation and differentiation of fibroblasts are hallmarks of pulmonary fibrosis. These cells are responsible for the excessive production of collagen and other extracellular matrix components, leading to tissue remodeling and scarring. Understanding the signals that drive this aberrant behavior is critical [4].
Beyond the TGF-β pathway, other signaling cascades like the Wnt/β-catenin pathway have been implicated in the development of pulmonary fibrosis. This pathway can promote fibroblast activation and contribute to the excessive synthesis of extracellular matrix, further exacerbating fibrotic changes [5].
Similarly, the Hippo signaling pathway, a critical regulator of organ size and cell proliferation, is also dysregulated in pulmonary fibrosis. Its abnormal functioning can lead to increased activity of downstream effectors that promote fibrotic processes [6].
Epithelial-mesenchymal transition (EMT) of alveolar epithelial cells represents another pivotal event in the pathogenesis of pulmonary fibrosis. During EMT, epithelial cells acquire mesenchymal characteristics, contributing to fibroblast activation and the deposition of extracellular matrix, thereby driving fibrosis [7].
The extracellular matrix (ECM) itself is not merely a passive scaffold but actively participates in regulating cellular behavior during fibrosis. Its abnormal deposition and remodeling significantly influence the fibrotic process and the progression of lung disease [8].
Therapeutic interventions targeting these molecular pathways are actively being investigated. Small molecule inhibitors, such as pirfenidone and nintedanib, have demonstrated efficacy in slowing disease progression, highlighting the importance of targeting specific fibrotic cascades [9].
Furthermore, the development of biologics, including antibodies or antagonists targeting key cytokines and growth factors like TGF-β and IL-13, represents another promising avenue for treating pulmonary fibrosis by modulating the immune and inflammatory responses driving fibrotic remodeling [10].
Pulmonary fibrosis is a formidable chronic lung disease characterized by the progressive scarring of lung tissue, leading to irreversible damage and impaired respiratory function. The underlying molecular mechanisms are intricate and involve a complex interplay of cellular signaling pathways and cellular interactions that ultimately result in the excessive deposition of extracellular matrix [1].
A central regulator of this fibrotic process is the Transforming Growth Factor-beta (TGF-β) signaling pathway. This pathway plays a pivotal role in driving fibroblast proliferation and promoting the differentiation of fibroblasts into myofibroblasts, which are critical for the excessive deposition of extracellular matrix components and the subsequent scarring of lung tissue [2].
The inflammatory milieu within the lung is a significant contributor to the pathogenesis of pulmonary fibrosis. Mediators such as Interleukin-13 (IL-13) and Tumor Necrosis Factor-alpha (TNF-α) are implicated in the chronic inflammation that fuels fibroblast activation and extracellular matrix accumulation, thereby exacerbating the fibrotic process [3].
Fibroblast activation and their subsequent differentiation into myofibroblasts are considered hallmarks of pulmonary fibrosis. These activated cells are primarily responsible for the excessive production of collagen and other extracellular matrix proteins, which leads to tissue remodeling and the characteristic scarring observed in fibrotic lungs [4].
In addition to the well-established TGF-β pathway, other signaling cascades, including the Wnt/β-catenin pathway, have been identified as significant contributors to the pathogenesis of pulmonary fibrosis. This pathway can potentiate fibroblast activation and enhance the synthesis of extracellular matrix, further driving fibrotic changes [5].
The Hippo signaling pathway, a highly conserved kinase cascade that regulates cell proliferation and organ size, is also implicated in the development of pulmonary fibrosis. Dysregulation of this pathway can lead to uncontrolled cell growth and contribute to fibrotic tissue remodeling [6].
Epithelial-mesenchymal transition (EMT) of alveolar epithelial cells represents another critical event in the development of pulmonary fibrosis. During EMT, epithelial cells undergo a transformation that promotes fibroblast activation and extracellular matrix deposition, playing a substantial role in the fibrotic process [7].
The extracellular matrix (ECM) is not merely a passive product of aberrant deposition but actively participates in modulating cellular behaviors during fibrosis. Its dynamic remodeling and composition significantly influence the progression of lung fibrosis and tissue scarring [8].
Given the complex molecular underpinnings, therapeutic strategies are being developed to target these pathways. Small molecule inhibitors, such as pirfenidone and nintedanib, have shown promise in clinical trials by effectively slowing the progression of idiopathic pulmonary fibrosis, underscoring the importance of targeting key signaling molecules [9].
Furthermore, biologics, including antibodies and antagonists designed to neutralize key cytokines and growth factors involved in fibrosis, such as anti-TGF-β antibodies and IL-13 antagonists, are under investigation. These approaches aim to modulate the immune and inflammatory responses that drive fibrotic remodeling in the lung [10].
Pulmonary fibrosis is a chronic lung disease marked by progressive lung tissue scarring, driven by complex molecular pathways. Key mechanisms include the TGF-β signaling cascade, inflammatory responses mediated by cytokines like IL-13 and TNF-α, and aberrant fibroblast activation into myofibroblasts. Dysregulation of Wnt/β-catenin and Hippo signaling pathways also contributes to the fibrotic process. Epithelial-mesenchymal transition (EMT) and the extracellular matrix's role in regulating cellular behavior are significant factors. Therapeutic strategies are being developed, including small molecule inhibitors like pirfenidone and nintedanib, and biologics targeting cytokines and growth factors, aiming to halt or reverse lung scarring.
None
None
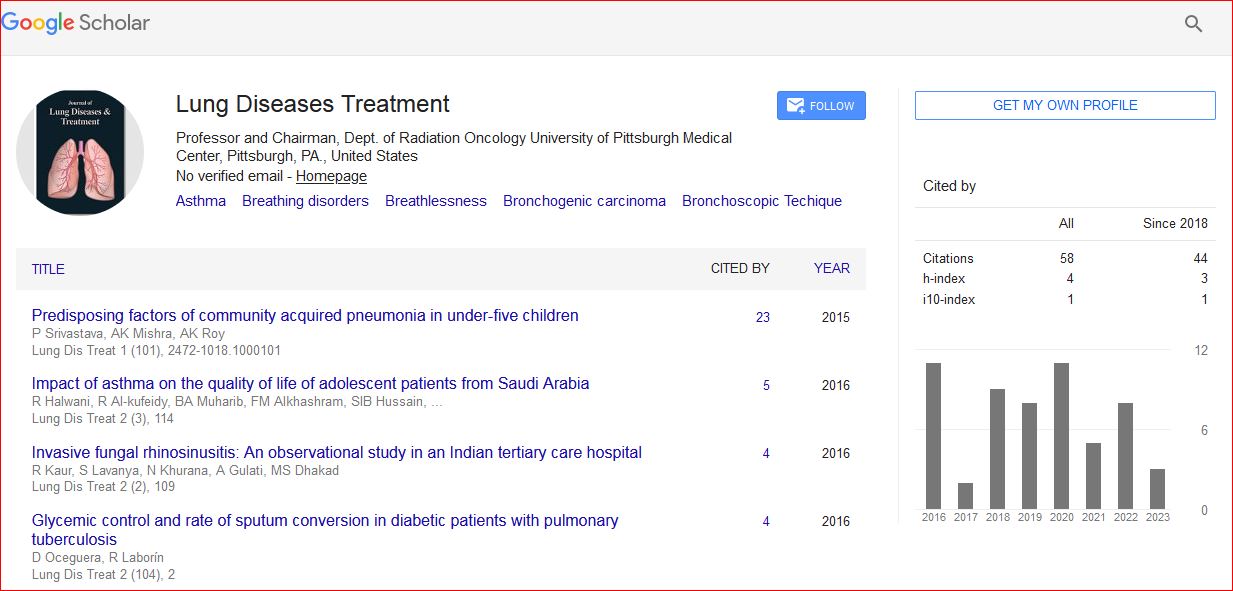
Journal of Lung Diseases & Treatment received 247 citations as per Google Scholar report