Opinion - (2025) Volume 10, Issue 3
Received: 02-Jun-2025, Manuscript No. jcct-26-183213;
Editor assigned: 04-Jun-2025, Pre QC No. P-183213;
Reviewed: 18-Jun-2025, QC No. Q-183213;
Revised: 23-Jun-2025, Manuscript No. R-183213;
Published:
30-Jun-2025
, DOI: 10.37421/2577-0535.2025.9.311
Citation: Kovacs, Natalia. ”Prioritizing Patient Safety in Oncology Trials.” J Cancer Clin Trials 10 (2025):311.
Copyright: © 2025 Kovacs N. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Ensuring patient safety is a paramount consideration in the complex landscape of oncology clinical trials. This multifaceted approach necessitates a rigorous assessment of drug toxicity, meticulous patient selection, and ongoing, vigilant monitoring for any adverse events that may arise during the course of treatment. A deep understanding of the potential risks associated with investigational therapies, coupled with the implementation of effective management strategies, is absolutely crucial for the ethical and successful conduct of these trials. Ultimately, the data and insights gleaned from these trials are instrumental in informing future clinical practice and guiding regulatory decisions for drug approval processes, representing a critical step in advancing cancer care [1].
The initial phases of oncology clinical trials, particularly Phase I studies, place a significant emphasis on the identification and thorough characterization of dose-limiting toxicities (DLTs). Establishing the maximum tolerated dose (MTD) is a fundamental objective that requires careful and comprehensive evaluation of both patient-reported outcomes and objective clinical assessments of toxicity. This intricate process is inherently iterative, relying heavily on the robust collection, precise interpretation, and diligent analysis of gathered data to effectively guide the subsequent phases of trial development and ensure patient safety at every step [2].
In the ever-evolving field of oncology, predictive biomarkers have emerged as increasingly vital tools for stratifying patient populations within clinical trials and for anticipating potential toxicities before they manifest. A profound understanding of the molecular underpinnings that govern drug response and resistance mechanisms allows for the development and application of more targeted therapies. This precision in treatment selection can potentially lead to a reduction in off-target effects, thereby enhancing both the overall efficacy of the treatment and its safety profile for the individual patient [3].
Complementing the data generated from traditional randomized controlled trials (RCTs), real-world data (RWD) and real-world evidence (RWE) are progressively becoming essential components in the comprehensive evaluation of oncology drugs. RWD, collected from routine clinical practice, can offer invaluable insights into the long-term safety and toxicity profiles of therapies when administered to broader and more diverse patient populations. This type of evidence plays a crucial role in post-marketing surveillance and provides critical information for regulatory decision-making processes [4].
The advent and widespread adoption of novel therapeutic modalities, such as immunotherapy and advanced cell-based therapies, have introduced unique and complex toxicity profiles that demand specialized approaches to monitoring and management. A thorough understanding of immune-related adverse events (irAEs) and the development of effective management strategies are absolutely critical for the safe and successful integration of these groundbreaking treatments into standard clinical practice, offering new hope for patients with various cancers [5].
Patient-reported outcome measures (PROMs) serve as indispensable tools for capturing the subjective experiences of patients regarding treatment-related toxicity and the overall burden of therapy. The systematic integration of PROMs into the routine conduct of clinical trials provides a more holistic and comprehensive understanding of drug safety from the patient's perspective. This patient-centric data effectively complements objective clinical assessments, offering a richer and more nuanced view of treatment impact [6].
An ongoing and critical area of research within oncology trials involves the development of novel endpoints that can more accurately and efficiently assess both treatment efficacy and toxicity. The utilization of dynamic and adaptive trial designs holds significant promise for improving the overall efficiency of clinical investigations. These designs allow for the earlier detection of potential safety signals and facilitate more rapid and informed decision-making throughout the trial process, ultimately benefiting future patient cohorts [7].
Established regulatory frameworks, meticulously developed and overseen by agencies such as the Food and Drug Administration (FDA) and the European Medicines Agency (EMA), provide essential and authoritative guidance for the design and rigorous conduct of oncology clinical trials. These guidelines place a particularly strong emphasis on comprehensive safety assessment, ensuring that adherence to these directives upholds the integrity of collected data and, most importantly, guarantees the protection of all trial participants throughout their involvement [8].
Pharmacovigilance, the science and activities relating to the detection, assessment, understanding, and prevention of adverse effects or any other medicine-related problem, plays a dynamic and indispensable role in oncology trials. This process involves continuous monitoring and diligent reporting of adverse events. The early detection of potential toxicities and their timely and appropriate management are not only critical for the immediate well-being of patients but also for the sustained and progressive advancement of drug development initiatives [9].
The ethical considerations that surround patient safety and toxicity in the context of oncology clinical trials are inherently multifaceted and require careful navigation. Striking an appropriate balance between the potential therapeutic benefits offered by novel treatments and the unavoidable inherent risks necessitates transparent and open communication with patients. This, combined with robust informed consent processes and an unwavering commitment to minimizing patient harm, forms the bedrock of ethical research conduct [10].
The fundamental principle guiding oncology clinical trials is the unwavering commitment to patient safety, which encompasses a comprehensive framework of rigorous toxicity assessment, judicious patient selection, and continuous, diligent monitoring for adverse events. Understanding the inherent risks associated with novel agents and proactively implementing effective management strategies are not merely procedural steps but are integral to conducting ethically sound and scientifically successful trials. The insights derived from these meticulously conducted studies are pivotal in shaping subsequent clinical practice and informing the critical processes of drug approval, thereby advancing the standard of care for cancer patients worldwide [1].
In the early stages of cancer drug development, particularly during Phase I trials, the identification and detailed characterization of dose-limiting toxicities (DLTs) are of paramount importance. The process of establishing a maximum tolerated dose (MTD) involves a careful and systematic evaluation of patient-reported outcomes, alongside objective assessments of observed toxicities. This iterative process is heavily dependent on the collection of high-quality data and its accurate interpretation, which collectively serve to inform and guide the progression to subsequent trial phases, ensuring that safety remains the foremost priority [2].
Predictive biomarkers are increasingly recognized for their significant role in stratifying patients for enrollment in oncology trials and for their utility in anticipating potential adverse reactions. A deeper understanding of the molecular mechanisms that underlie drug response and resistance allows for the design of more targeted therapeutic strategies. This targeted approach has the potential to minimize off-target effects, leading to improved efficacy and enhanced safety profiles for patients receiving these novel treatments [3].
The integration of real-world data (RWD) and the subsequent generation of real-world evidence (RWE) are becoming indispensable supplements to the data obtained from traditional randomized controlled trials (RCTs) in oncology drug development. RWD can provide invaluable longitudinal insights into the safety and toxicity profiles of drugs when used in broader and more heterogeneous patient populations, thereby significantly aiding in post-marketing surveillance and supporting regulatory decision-making processes [4].
As novel therapeutic modalities such as immunotherapy and cell-based therapies continue to gain prominence, they bring with them unique toxicity profiles that necessitate specialized monitoring and management strategies. A thorough understanding of immune-related adverse events (irAEs), including their mechanisms and effective management, is crucial for the safe and successful implementation of these innovative and potentially life-saving treatments into clinical practice [5].
Patient-reported outcome measures (PROMs) are essential instruments for capturing the subjective experiences of patients concerning treatment toxicity and the overall burden of therapy. Incorporating PROMs into the standard procedures of clinical trials provides a more complete and patient-centered understanding of drug safety. This data effectively complements objective clinical assessments, offering a more holistic view of the treatment's impact on patients' lives [6].
The development of innovative endpoints for assessing both the efficacy and toxicity of treatments in oncology trials is a dynamic and active area of research. The implementation of adaptive trial designs offers the potential to enhance trial efficiency, enabling earlier identification of safety signals and facilitating more timely decision-making. This can lead to quicker advancements in drug development and a more rapid availability of effective treatments for patients [7].
Regulatory frameworks established by leading health authorities, such as the FDA and EMA, provide critical guidance for the design and execution of oncology clinical trials, with a pronounced emphasis on comprehensive safety evaluations. Strict adherence to these regulatory guidelines is imperative for ensuring the scientific integrity of the collected data and, most importantly, for safeguarding the well-being and rights of all participants involved in the trials [8].
Pharmacovigilance in oncology clinical trials is an ongoing and dynamic process that requires continuous monitoring and prompt reporting of all adverse events. The early detection and appropriate management of toxicities are of utmost importance for ensuring patient safety and for the successful and progressive development of new anti-cancer drugs [9].
Ethical considerations concerning safety and toxicity in oncology trials are complex and demand careful deliberation. The process of balancing the potential benefits of investigational treatments with their inherent risks requires open and honest communication with patients, a robust informed consent process, and a steadfast commitment to minimizing any potential harm experienced by trial participants [10].
Oncology clinical trials prioritize patient safety through rigorous toxicity assessment, careful patient selection, and continuous monitoring for adverse events. Understanding potential risks and implementing effective management strategies are crucial for ethical and successful trial conduct, informing clinical practice and drug approval. Dose-limiting toxicities (DLTs) and maximum tolerated doses (MTDs) are fundamental in early-phase trials, requiring robust data collection and interpretation. Predictive biomarkers help stratify patients and anticipate toxicities by understanding molecular responses. Real-world data (RWD) and evidence (RWE) supplement trial data, offering insights into long-term safety in broader populations. Novel therapies like immunotherapy present unique toxicity profiles necessitating specialized monitoring. Patient-reported outcome measures (PROMs) capture subjective patient experiences of toxicity, complementing objective assessments. Adaptive trial designs aim to improve efficiency by enabling earlier detection of safety signals. Regulatory frameworks provide essential guidance for trial conduct and safety assessment. Pharmacovigilance involves ongoing monitoring and reporting of adverse events to ensure patient well-being and drug development progress. Ethical considerations require balancing potential benefits with inherent risks, emphasizing transparent communication and informed consent.
None
None
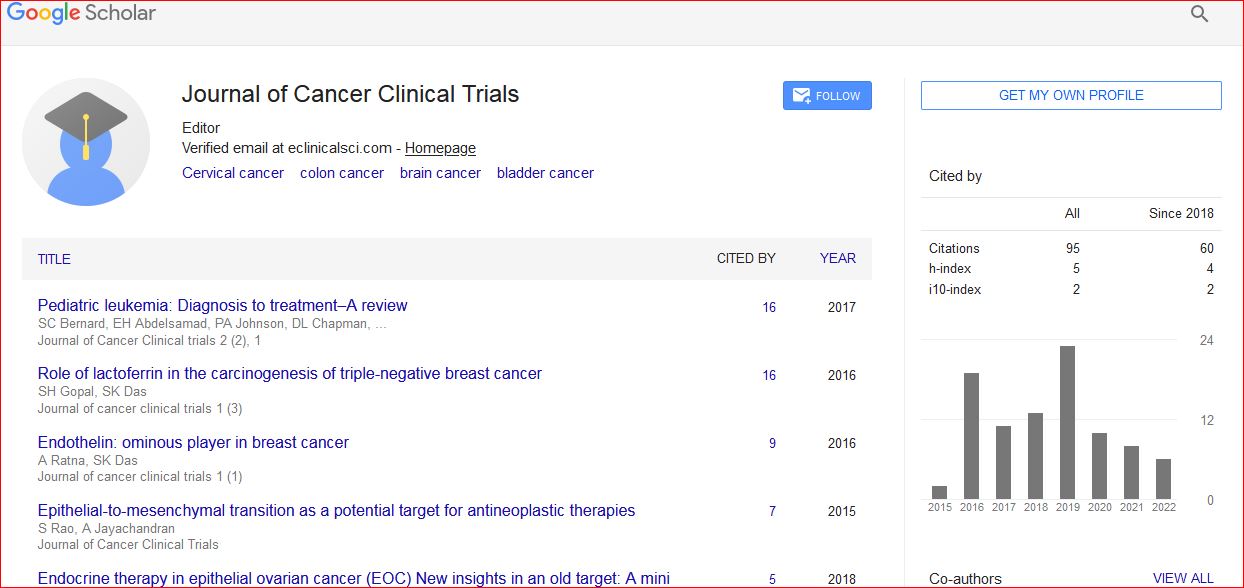
Journal of Cancer Clinical Trials received 95 citations as per Google Scholar report