Editorial - (2021) Volume 5, Issue 5
Received: 08-Sep-2021
Published:
20-Sep-2021
, DOI: 10.37421/jmbp.2021.5.134
Citation: Sandeep Kaur Ravala. "Molecular Docking:
Bioinformatics Tool." J Microbiol Pathol 5 (2021): 134.
Copyright: © 2021 Sandeep Kaur Ravala. This is an open-access article
distributed under the terms of the Creative Commons Attribution License, which
permits unrestricted use, distribution, and reproduction in any medium, provided
the original author and source are credited.
Molecular docking analysis has been one among the foremost basic and important strategy for drug discovery. Many diseases are caused by the malfunction of proteins and therapies are focused on the inhibition or activation of the target proteins. It allows prediction of molecular interactions that hold together a protein and a ligand within the bound state. During this chapter, a quick introduction of molecular docking, alongside the available methods, their development and application in drug discovery has been presented. The relevant basic theories, including sampling algorithms and scoring functions, also are mentioned. Comparative analysis of various molecular docking approaches, especially that including backbone flexibility in receptors, has also been included within the discussion. Considering the importance of application of such tools and methods, a solved practical exercise alongside an in depth outline of the protocol to follow, has been provided within the final section of the chapter. All the calculations are performed using free-wares like Auto Dock, so any and each reader can practice and validate their docking study. The classical mechanism of computer-based drug designing methods masks the function of person proteins of interest. As a comprehensive approach, for the treatment of Parkinson and a number of other inflammatory disorders. In molecular modeling, docking may be a method which predicts the well-liked orientation of 1 molecule to a second when sure to one another to make a stable complex. Knowledge of the orientation successively could also be wont to predict the strength of association or binding affinity between two molecules using, for instance, scoring functions. Schematic illustration of docking a little molecule ligand (green) to a protein target (black) producing a stable complex.
The associations between biologically relevant molecules like proteins, peptides, nucleic acids, carbohydrates, and lipids play a central role in signal transduction. Furthermore, the relative orientation of the 2 interacting partners may affect the sort of signal produced (e.g., agonism v/s antagonism). Therefore, docking is beneficial for predicting both the strength and sort of signal produced. Molecular docking is one among the foremost frequently used methods in structure-based drug design, thanks to its ability to predict the bindingconformation of small molecule ligands to the acceptable target binding site. Characterizations of the binding behavior play a crucial role in rational design of medicine also on elucidate fundamental biochemical processes.
Two approaches are particularly popular within the molecular docking community. One approach uses an identical technique that describes the protein and therefore the ligand as complementary surfaces. The second approach simulates the particular docking process during which the ligand-protein pairwise interaction energies are calculated. Both approaches have significant advantages also as some limitations. These are outlined below.
Shape complementarity
Geometric matching/ shape complementarity methods describe the protein and ligand as a group of features that make them dock able. These features may include molecular surface / complementary surface descriptors. During this case, the receptor's molecular surface is described in terms of its solvent-accessible area and therefore the ligand's molecular surface is described in terms of its matching surface description. The complementarity between the 2 surfaces amounts to the form matching description which will help finding the complementary pose of docking the target and therefore the ligand molecules. Another approach is to explain the hydrophobic features of the protein using turns within the main-chain atoms. Yet one more approach is to use a Fourier shape descriptor technique. Whereas the form complementarity based approaches are typically fast and robust, they can't usually model the movements or dynamic changes within the ligand/ protein conformations accurately, although recent developments allow these methods to research ligand flexibility. Shape complementarity methods can quickly scan through several thousand ligands during a matter of seconds and truly find out whether or not they can bind at the protein's site, and are usually scalable to even protein-protein interactions. They’re also far more amenable to pharmacophore based approaches, since they use geometric descriptions of the ligands to seek out optimal binding. Molecular docking may be a simplified sort of MD simulation. It is often used on intervals to exchange lengthy segments of MD simulation trajectories, especially in cases where certain domains undergo large translations, rotations, and conformation changes.
Simulation
Simulating the docking process is far more complicated. During this approach, the protein and therefore the ligand are separated by some physical distance, and therefore the ligand finds its position into the protein's site after a particular number of “moves” in its conformational space. The moves incorporate rigid body transformations like translations and rotations, also as internal changes to the ligand's structure including torsion angle rotations. Each of those moves within the conformation space of the ligand induces a complete energetic cost of the system. Hence, the system's total energy is calculated after every move. Simulation is computationally expensive, having to explore an outsized energy landscape. Grid-based techniques, optimization methods, and increased computer speed have made docking simulation more realistic.
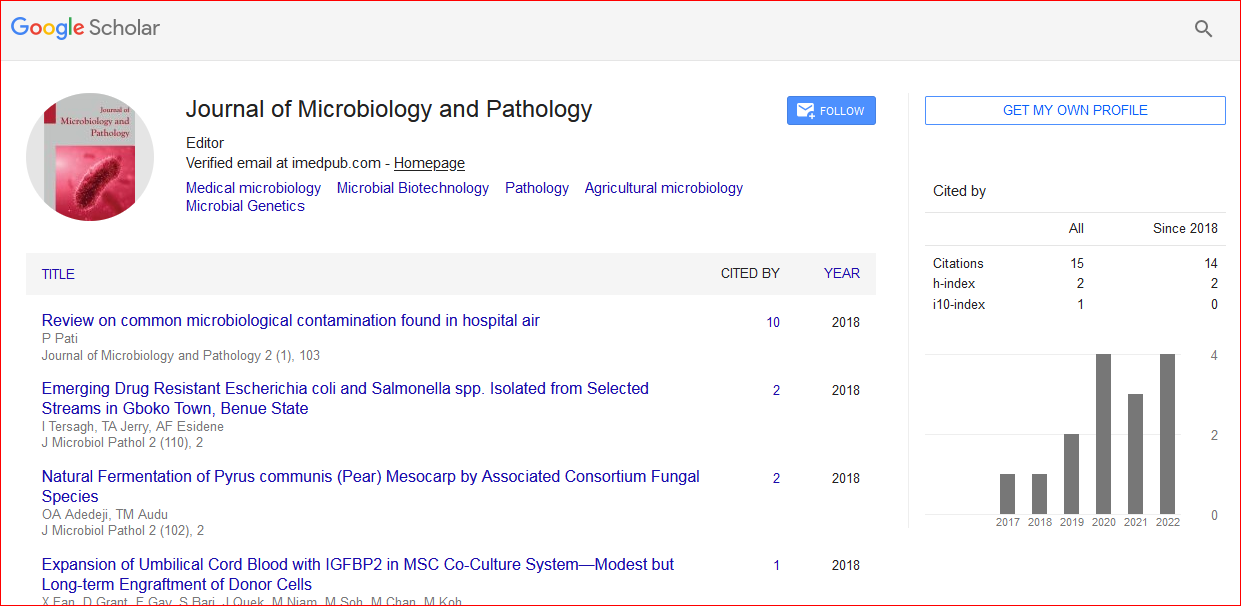
Journal of Microbiology and Pathology received 18 citations as per Google Scholar report