Editorial - (2022) Volume 13, Issue 4
Received: 04-Apr-2022, Manuscript No. CSJ-22-65459;
Editor assigned: 05-Apr-2022, Pre QC No. P-65459;
Reviewed: 18-Apr-2022, QC No. Q-65459;
Revised: 19-Apr-2022, Manuscript No. R-65459;
Published:
28-Apr-2022
, DOI: 10.37421/2150-3494.2022.13.287
Citation: Kenny, Petter. “Intramolecular Aminoarylation of Unactivated Alkenes with Aryl Sulfonamides Catalysed by Aryl Sulfonamides” Chem Sci J 13 (2022): 287.
Copyright: © 2022 Kenny P. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
The arylethylamine pharmacophore is saved across a scope of organically dynamic regular items and medications, especially in particles that follow up on the focal sensory system Conventional arrangements of arylethylamines depend on straight, stoichiometric changes to fashion key C and C-N bonds. Such courses miss the mark on combinatorial adaptability leaned toward in beginning phase restorative science missions and they limit the available replacement examples of the ethylene linker piece. Substituents on the linker can definitely change the particle's lipophilicity, adaptation, and end half-life. Modular arrangements of complex arylethylamines from industrially accessible or handily integrated substrates are in this way profoundly important [1].
The main case depicts hostile to Markovnikov hydroamination of a styrene, and numerous techniques exist to achieve this change really with the guide of photoredox, lanthanide, or progress metal catalysts. The subsequent case requires against Markovnikov hydroarylation of an enamine, which was as of late detailed in great yields by Jui and co-workers. The third case involves aminoarylation of an unactivated alkene and is, on a basic level, the most particular of the three difunctionalization methodologies. Since the substrate is decoupled from both the arene and the nitrogen particle, straightforward alkenes can be changed over completely to arylethylamines in one stage. Our inclinations in complex particle blend by revolutionary strategies drove us to address whether aminoarylation could be accomplished with nitrogenfocused extremists [2]. Such electrophilic intermediates can be gotten to promptly through photoredox catalysis. We saw the advances by Knowles and colleagues in synergist N-focused revolutionary age as especially empowering towards our goal.
Formal homolysis of N-H bonds by means of various site coordinated proton-electron move (MS-CPET) licenses valuable reactivity of N-focused extremists without the requirement for cruel oxidants or solid bases. If the N-H bond is adequately acidic, stepwise deprotonation/oxidation successions can likewise give N-focused revolutionaries under gentle conditions.
We originally viewed as the cutting edge in unactivated alkene aminoarylation to illuminate our response plan. Palladium-catalyzed alkene aminoarylation was investigated broadly by Wolfe and collaborators in the planning of soaked nitrogen heterocycles. The Chemler and Antonchik bunches have advanced copper-catalyzed and metal-free ways to deal with intramolecular carboamination. Engle and associates utilized guiding gatherings to organize palladium-and nickel-catalyzed intermolecular aminoarylations of β,γ-unsaturated enamides and of homoallylic alcohols, respectively. The Molander and Leonori labs blended photoredox-and progress metal catalysis by catching amidyl revolutionary cyclization intermediates with nickel to achieve C cross-coupling. Gaunt and collaborators accomplished a three-part copper-catalyzed alkene azidoarylation utilizing diaryliodonium triflates as aryl extremist precursors. We imagined a desulfonylative 1,4-aryl relocation (Smiles-Truce reworking) as an unusual disengagement of the C security that could be initiated by a N-focused sulfonamidyl revolutionary expansion to an alkene [3,4].
Aryl movements have become progressively open to manufactured physicists as present day photochemical procedures have matured. For this situation, the relocation would give passage to the arylethylamine platform from reasonable sulfonamides. Mechanistically, this particular fountain wouldn't need a cross-coupling impetus and may concede admittance to sterically clogged items that are trying to plan through change metal-interceded techniques. Despite the fact that Molander's way to deal with aminoarylation starts by a N-focused extremist cyclization from a N-H security, the MS-CPET technique decided to produce the revolutionary requires N-aryl amide forerunners [5].
Oxidative cleavage of the helper arene in the item is subsequently important to give the free lactam. On the other hand, the planned desulfonylative aryl relocation in this work would work as an in situ deprotection of the nitrogen molecule. We recently detailed an alkene aminoarylation that continued through alkene revolutionary cation intermediates. These electrophilic species effectively combined with sulfonamides, prompting a Smiles-Truce revamp that conveyed the ideal arylethylamine. Notwithstanding, just electron-rich, 1,2-disubstituted styrenes gave great yields. This limitation was ascribed to the low oxidation possibilities of the initiated alkenes and the higher affinity of monosubstituted styrene extremist cations to oligomerize. We expected our N-focused revolutionary way to deal with dodge this impediment too, in view areas of strength for of point of reference portraying against Markovnikov sulfonamidyl extremist augmentations to unactivated alkenes.
None.
The authors declare that there is no conflict of interest associated with this manuscript.
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
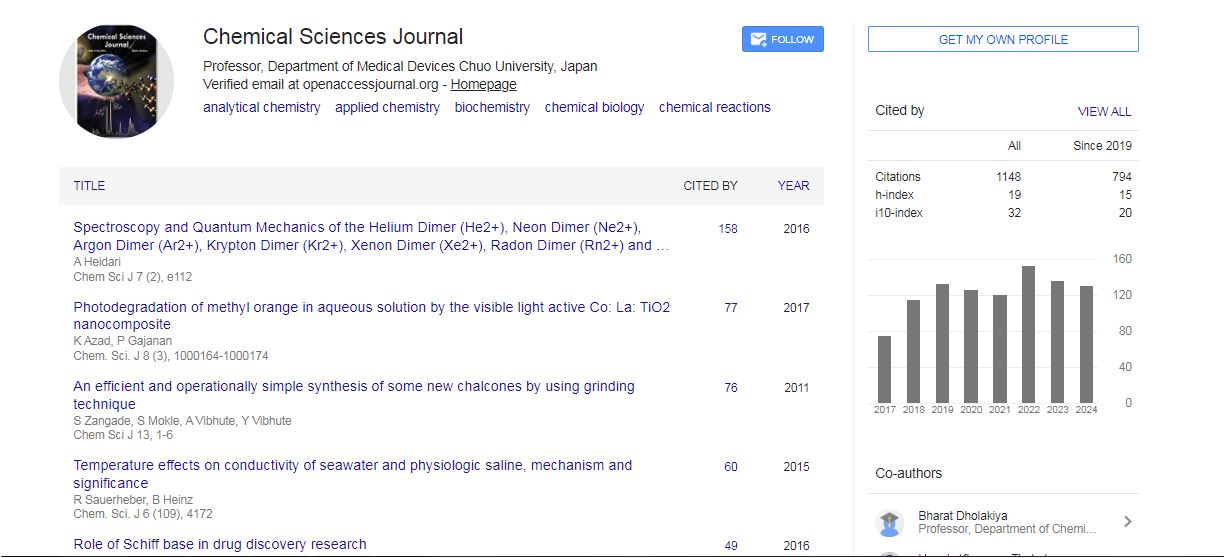
Chemical Sciences Journal received 912 citations as per Google Scholar report