Research Article - (2022) Volume 14, Issue 3
Received: 10-Mar-2022, Manuscript No. jcst-22-56734;
Editor assigned: 14-Mar-2022, Pre QC No. P-56734;
Reviewed: 21-Mar-2022, QC No. Q-56734;
Revised: 24-Mar-2022, Manuscript No. R-56734;
Published:
31-Mar-2022
, DOI: 10.37421/1948-5956.22.14.519
Citation: Chu, Fei, Nicholas J. Skertich, Timothy B. Lautz and Stephen Szajek, et al. “HDAC-6 Regulation of HIF-1α Plays an Important Role in Mediating both Drug Resistance and Invasiveness in Osteosarcoma and Breast Cancer Cells.” J Cancer Sci Ther 14 (2022): 519. DOI: 10.37421/1948-5956.22.14.519.
Copyright: © 2022 Chu F, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Sources of funding : John W. Anderson Foundation (Valparaiso, IN), the Super Jake Foundation (Gurnee, IL) and the Michael Reese Research and Education Foundation (Chicago, IL), Beckley Foundation (Chicago, IL) and Alex Manderino Foundation (St Joseph, MI).
Objective: Despite advances in cancer treatment, chemotherapy resistance and metastasis are major hurdles in curative cancer treatment. Studies involving drug resistance and cancer metastasis have generally proceeded along separate pathways of research. Current interest has focused on a possible relationship between drug resistance and cancer metastasis, since the molecular basis for drug resistance with an aggressive metastatic phenotype remains to be elucidated. We aim to show that histone deacetylase (HDAC) involvement in hypoxia-inducible factor-1α (HIF-1α) regulation may connect drug resistance to cancer invasiveness in osteosarcoma and breast cancer.
Methods: We created a doxorubicin resistant (DoxR) cell line from wildtype (WT) human osteosarcoma (SJSA-1) and breast cancer (MCF-7) cells. Matrigel in vitro invasion assay and colony formation assay were used to compare invasiveness of WT to DoxR cells. Western blot assay was used to determine HDAC and HIF-1α expression. Invasiveness and expression of HDACs and HIF-1α between WT and DoxR cells were also compared after treatment with vorinostat, an HDAC inhibitor, and in small interfering RNA (siRNA)-mediated knockdowns of HDAC-6 and HIF-1α.
Results: Both DoxR SJSA-1 and MCF-7 cells were more invasive than their doxorubicin-sensitive WT cells. Expression of HDAC-6 and HIF- 1α was increased in DoxR compared to WT cells. Inhibition of HDAC-6, either by the HDAC inhibitor (HDACi) vorinostat or small interfering RNA (siRNA)-mediated knockdown, decreased HIF-1α expression and the invasiveness of DoxR cells. Small hairpin RNA (shRNA)-mediated knockdown of HIF-1α also suppressed the invasiveness of DoxR cells and sensitized the DoxR Cell to doxorubicin.
Conclusion: These findings demonstrate that HDAC6 is involved in the regulation of HIF-1α and might connect drug resistance and cancer invasion.
Doxorubicin • Drug resistance • Cancer invasiveness • Histone deacetylases (HDACs) • Hypoxia Inducible factor-1 α (HIF-1 α) • Osteosarcoma (SJSA-1) • Breast cancer (MCF-7)
Glossary of Abbreviations: MDR: Multi-Drug Resistance; HIF-1α: Hypoxia-inducible Factor 1α; HDACi: Histone Deacetylase Inhibitor; HDAC: Histone Deacetylase
Despite advances in cancer treatment, chemotherapy resistance and metastasis are major barriers to cure [1,2]. Historically, the study of drug resistance and metastasis have proceeded along separate pathways [3,4]. However, growing evidence has demonstrated that chemoresistance and invasiveness may be linked [4,5]. We recently demonstrated that neuroblastoma cells display characteristics of mesenchymal change via multiple pathways in their transition to a drug-resistant state [6].
The tumor microenvironment is ever-changing and experiences fluctuation in hypoxia and nutrient deprivation that can lead to epigenetic and genetic adaptations that increase invasiveness and metastasis [7]. Study of the regulation of gene products in response to hypoxia has found the transcription factor hypoxia-inducible factor 1α (HIF-1α) to be involved. HIF- 1α is consistently implicated in cancer metastasis and its overexpression is a marker of poor prognosis [7-9]. This holds true in osteosarcoma and breast cancer as well [7,10,11]. It is involved in the activation of numerous cellular processes including resistance against apoptosis, over-expression of drug efflux membrane pumps, vascular remodeling, angiogenesis and metastasis, making it a target for cancer therapy research [4,8,12].
Accumulating evidence suggests that HIF-1α activity in tumor cells can be repressed by histone deacetylase inhibitors (HDACIs) [13-15]. Histone deacetylases (HDACs) are important epigenetic regulators of gene transcription and play a role in tumorigenesis [2,16-18]. In our prior work on doxorubicin resistant neuroblastoma cells, we showed that HDACs play a significant role in drug resistance [19,20]. HDACIs have demonstrated a wide range of effects on cancer cells, including growth inhibition, induction of cell death, and antiangiogenesis [2,6,21]. Vorinostat is the first FDA-approved HDACI and is currently in phase I and II clinical trials for a number of hematologic and solid organ malignancies [22]. HDACIs have been shown to have in vitro success in various osteosarcoma and breast cell lines, but have yet to be linked to HIF-1α expression suppression [23,24]. Given the role of HIF-1α in cancer metastasis and its regulation by HDACs, which have also been shown to play a role in chemoresistance, we hypothesize that HDAC-mediated control of HIF- 1α activity functions as a link between chemoresistance and invasiveness in doxorubicin resistant osteosarcoma and breast cancer cells.
Reagents
Human osteosarcoma (SJSA-1) and embryonic kidney (HEK 293T/17) cell lines were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). The human breast adenocarcinoma (MCF7) cell line was obtained from Dr. William S. Dalton, PhD, MD (H. Lee Moffitt Cancer Center & Research Institute, FL, USA). Dulbecco’s modified Eagle’s medium (DMEM) and fetal bovine serum (FBS) were obtained from Corning (Corning, NY, USA). Penicillin and streptomycin were obtained from HyClone (Logan, UT, USA). Plasmocin prophylactic was obtained from InvivoGen (San Diego, CA, USA). Vorinostat was purchased from ChemieTek (Indianapolis, IN, USA). Doxorubicin, 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide (MTT), and deferoxamine mesylate (DFO), were purchased from Sigma- Aldrich (St. Louis, MO, USA). The following primary antibodies were obtained: HDAC-6 (Santa Cruz Biotechnology, Dallas, TX, USA, sc-28386), HIF-1α (BD Biosciences, Franklin Lakes,NJ,USA, 610958) and β-actin (Sigma- Aldrich, A5441). Secondary antibodies conjugated to horseradish peroxidase (HRP) were purchased from Promega (Madison, WI, USA, goat anti-mouse immunoglobulin G (IgG)-HRP, W402B; goat anti-rabbit IgG-HRP, W401B). Enhanced chemiluminescence reagents, iBlot 2 and transfer mini stacks for Western blots were obtained from Thermo Fisher Scientific (Asheville, NC, USA).
Cell culture, drug treatment and cytotoxicity assay
Cells were cultured either in normal conditions (37°C and 5% CO2 equilibrated with atmospheric O2 in a humidified incubator) that contains 21% O2 (hereafter referred as normoxia) or in the hypoxia chamber (STEMCELL Technologies, Vancouver, Canada, Cat. No.27310, 1% O2, 5% CO2, balanced with N2 and humidified) that was placed in 37°C (hereafter referred as hypoxia). Cells were cultured in complete medium (DMEM supplemented with 10% heat-inactivated FBS, 100 units/mL penicillin, 100 μg/mL streptomycin and 5 μg/mL plasmocin). Doxorubicin-resistant (DoxR) cells were generated by incubating parental WT cells with incremental concentrations of doxorubicin ranging from 1 × 10-9 to 10-6 M over a six month period. Cells were considered to be DoxR after surviving five consecutive passages in 1 μM doxorubicin. Cell viability was determined by the quantitative colorimetric MTT assay according to Boehringer Mannheim (Cat. No. 1465 007) as previously described [25]. Briefly, cells were seeded in complete medium at 5 × 103 cells/well in 96-well plates and maintained in culture for 24 h at 37°C. Doxorubicin was added to designated wells at final concentrations of 1 × 10-9 to 10-5 M and incubated for 96 h at 37°C. The MTT reagent was added at a final concentration of 0.5 mg/ mL and incubated for 4 h at 37°C. Cells were solubilized for 16 h at 37°C. The optical density of this solution was measured at 570 nm (EL X800 plate reader, BioTek, Winooski, VT, USA) and the percentage of viable cells was determined by comparison with untreated control cells.
In vitro invasion assay
Cell invasion was determined and analyzed using a membrane invasion culture system purchased from Fisher Scientific (Chicago, IL, USA). The number of cells able to invade through a membrane coated with the defined Matrigel extracellular matrix during a 24 h period was compared to the number counted using a control insert with no Matrigel. Cells were seeded at 2.5 × 104 and incubated for 24 h. Cells that migrated through the membrane were fixed and stained with a Diff‐Quik staining kit obtained from Electron Microscopy Sciences (Hatfield, PA, USA). Three fields at 40X magnification were counted by light microscopy (technical replicates) for each experiment. All experiments were repeated in triplicate and reported as the number of cells on the membrane divided by the number on the control membrane (mean ± standard error).
Anchorage-independent assay
A soft agar colony formation assay was performed to evaluate anchorageindependent growth. Both SJSA-1 and MCF-7 cells (3 × 104) were suspended in 0.35% agarose in complete medium and plated into six-well dishes on 0.7% agar in the same medium. Colonies grew and pictures were taken on day 7. After three weeks, colonies were stained with 0.5% crystal violet for 30 minutes at room temperature, and the number of colonies that exceeded 250 μm in diameter per unit field was determined using a low-power microscope. Seven fields were counted to obtain the average number of colonies and the experiment was repeated thrice.
Colony formation assay
SJSA-1 and MCF-7 cells were plated at 3 x 103 per well in six-well tissue culture plates (Costar, Corning, NY). Twenty-four hours after plating, cells were incubated in hypoxia or left in normoxia for another 7 days. Colonies were fixed with 70% methanol and stained with methylene blue and colonies of >50 cells were counted. The number of colonies/plate that of >50 cells are displayed graphically (means ± standard error of three plates).
SDS‐PAGE and Western Blot
Parental and DoxR cells were seeded in complete medium and cultivated for 48 h. Cells were lysed using NP40 Cell Lysis Buffer purchased from Thermo Fisher Scientific (Asheville, NC, USA) with Protease Inhibitor Cocktail obtained from Sigma‐Aldrich (St. Louis, MO, USA). Total protein concentration was determined using the bicinchoninic acid assay (BCA) assay from Thermo Fisher Scientific (Asheville, NC, USA) using the supplied albumin as the analytical standard. Equal amounts of protein were reduced in 1X sample buffer (Laemmli) from Bio‐Rad (Hercules, CA, USA), with 5% β‐mercaptoethanol from Fisher Scientific (Chicago, IL, USA) boiled for five minutes, separated by electrophoresis on 4–20% Mini‐Protean TGX Precast Protein Gels obtained from Bio‐Rad (Hercules, CA, USA) and transferred using the Invitrogen iBlot 2 Gel Transfer Device purchased from Thermo Fisher Scientific (Asheville, NC, USA), onto nitrocellulose membranes via iBlot 2 Transfer Stacks also purchased from Thermo Fisher Scientific (Asheville, NC, USA). Proteins of interest were identified with specific primary antibodies followed by HRPconjugated secondary antibodies. Immunoreactive bands were detected by chemiluminescence with image capture on an iBright CL 1500 Imaging System bought from Thermo Fischer Scientific (Asheville, NC, USA). Three separate Western blot experiments were conducted. When assessing for HIF-1α protein expression, HIF-1α protein was stabilized by treating cells with 250 μM deferoxamine (DFO) for 16 h prior to whole cell lysis. DFO stabilizes HIF-1α by preventing O2-dependent proteasomal degradation.
Immunoprecipitation
SJSA-1 whole cell lysates were prepared as described above and diluted to a concentration of 1 μg/μL total protein using the BCA assay. Lysate (100 μL) and HIF-1α primary antibody (10 μL) were incubated overnight at 4°C under gentle agitation. Protein G-coupled agarose beads (Abcam, MA, USA) were prepared by mixing 200 μL of beads with 400 μL of PBS. The bead slurry was added to the lysate-antibody mixture and incubated overnight at 4°C under gentle agitation. The beads were centrifuged and washed with PBS three times. Lysis buffer (80 μL) and 5X reducing sample buffer (20 μL) were added to the resin, the mixture was boiled for five minutes and centrifuged to pellet the resin. Equal volumes (40 μL) of the resultant supernatant were processed for immunoblotting as described above.
Small molecule and siRNA inhibition of HDACs
Class I and II HDAC activity was inhibited by treating cells with 1 μM vorinostat for 48 h before performing the in vitro invasion assay. Isoform-specific HDAC-6 knockdown was achieved using siRNA synthesized by (Dharmacon, Lafayette, CO, USA). On the day before transfection, 3 × 105 cells were seeded into six-well plates and grown in 2.5 mL of complete medium. After 24 h in culture, 25 μL of 20 μM stock solution of siRNA duplexes were transfected into cells using the GeneSilencer® siRNA Transfection Reagent according to the manufacturer’s protocol (Gene Therapy Systems, San Diego, CA, USA). Cells were maintained in culture for 72 h before determining expression of the silenced molecules by Western blot or performing the in vitro invasion assay.
Small hairpin RNA inhibition of HIF-1α
A validated 29mer shRNA construct (pGFP-V-RS) against human HIF- 1α and scramble sequence were purchased from OriGene (Rockville, MD. USA). A polyclonal population of stably transfected cells was used in the study. Packing cells (HEK 293T/17) were transfected according to the HuSH-29TM shRNA plasmid application guide (OriGene, Rockville, MD. USA). Transfected cells were incubated for 48 h at 37°C for virus production. The virus-containing medium was collected and filtered by 0.45μm low protein binding filter. Target cells (SJSA-1 DoxR cells) were infected by treatment with polybrene (4 μg/ mL) and incubation with virus-containing medium for 72 h. Target cells were then treated with DFO for 16 h to prevent HIF-1α degradation before whole cell lysates were obtained and analyzed by Western blot. Stable clones or mixed populations were cultured in the presence of puromycin (2 μg/mL).
Statistical analysis
For in vitro invasion assays, categorical variables were compared between groups using chi‐square tests. For Western blot analysis, differences between parental and DoxR cell lines were assessed using Student’s unpaired t‐tests. P values <0.05 were considered statistically significant, (*) indication of significance in the bar graphs. Analysis were performed using IBM SPSS Statistics for Windows, version 26 (IBM corp., Armonk, N.Y., USA).
Doxorubicin-resistant cells are more invasive than their parental WT cells
The Matrigel in vitro invasion assay was used to compare the invasiveness of human osteosarcoma SJSA-1 DoxR cells and breast adenocarcinoma MCF7 DoxR cells to their parental WT cell lines. Normalized invasion indices were significantly higher in resistant cell lines compared to their parental WT lines (fraction of invasion 0.275 versus 0.156, P <0.05 in SJSA-1 cells, as well as 0.147 versus 0.0413, P <0.01 in MCF-7 cells) (Figure 1A and B), the result confirmed our previous finding in human neuroblastoma cells. The soft agar assay was used to evaluate the anchorage-independent growth of human neuroblastoma SJSA-1 DoxR cells and MCF7 DoxR cells compared to their parental WT cells. As shown in Figure 1C, SJSA-1 DoxR cells had increased colony formation (189 ± 23 colonies) compared to their parental WT cells (72 ± 16 colonies; P <0.001). Likewise, colony formation in soft agar was increased in MCF7 DoxR cells (106 ± 12 colonies) compared to their parental WT line (29 ± 7 colonies, P < 0.001) (Figure 1D).

Figure 1. Doxorubicin-resistant (DoxR) cells are more invasive than their parental wild-type (WT) counterparts. The Matrigel in vitro invasion assay was used to compare DoxR cells to their parental WT cells in (A) SJSA-1, (B) and MCF7. Invasion was calculated as the percentage of cells able to invade through a membrane coated with Matrigel during a 24 h period as a fraction of the control. Bars represent the normalized invasion indices (mean ± standard error). Clonogenic survival of WT and DoxR cells were compared in a soft agar assay. Phase-contrast microscopy images of (C) SJSA-1 and (D) MCF7 cells cultured for 7 days in soft agar and stained with crystal violet are shown. The number of colonies/plate that exceeded 250 μm in diameter are displayed graphically (mean ± standard error of three plates), (*) indication of significance (P < 0.05).
Colony formation assay
Colony formation assay was used to evaluate the clonogenic ability of the SJSA-1 and MCF-7 cells WT and DoxR cells under both nomoxic and hypoxic conditions (Figures 2A-D). Results from three independent experiments showed that absolute clonogenicity (number of colonies/number of cells originally seeded) was significantly different between normoxic and hypoxic conditions, with significantly more colonies in the hypoxia treated groups than those in the normoxia controls in both cell lines (P <0.0001). Results also showed more spheres in SJSA-1 and MCF-7 DoxR cells than their parental WT counterparts under either normoxic or hypoxic conditions (Figures 2A-D).

Figure 2. Clonogenic survival of the WT and DoxR cells were compared. (A) SJSA-1 and (B) MCF-7 cells were plated at 3,000 per well in six-well tissue culture plates. Twenty-four hours after plating, cells were incubated in hypoxia or left in normoxia for another 7 days. Colonies were fixed with 70% methanol and stained with methylene blue and colonies of >50 cells were counted. Absolute clonogenicity was significantly different between normoxic and hypoxic conditions. The number of (C) SJSA-1 and (D) MCF-7 cell colonies/plate that are greater than 50 cells are displayed graphically (mean ± standard error of three plates). Data are representative of three independent experiments, (*) indication of significance (P < 0.05).
HDAC-6 is upregulated in doxorubicin resistant cells and plays a role in mediating HIF-1α activity
The protein level from whole cell lysates of HDAC-6 was upregulated in both DoxR SJSA-1 and MCF-7 cells when compared to their counterparts WT cells (Figure 3A). Likewise, after pre-treatment with DFO for 16 hours to prevent degradation of HIF-1α, HIF-1α levels were also increased in DoxR cells compared to their parental WT cell line (Figure 3B). HDAC inhibition with vorinostat for 48 hours prior to whole cell lysis reduced the level of HIF-1α protein in Dox R cells (Figure 3B). Knockdown of HDAC-6 with siRNA led to decreased HIF-1α expression as well (Figure 3D). Immunoprecipitation for HIF-1α was performed to identify specific HDAC isoforms that physically interact with this protein and demonstrated interaction with HDAC-6. Vorinostat pre-treatment reduced the level of HDAC-6 protein in the HIF-1α immunoprecipitated (Figure 3C).

Figure 3. Western blot demonstrating increased HDAC-6 expression in both SJSA-1 and MCF-7 DoxR cells compared to their WT counterparts (A), as well as increased HIF-1α expression in DoxR cells compared to WT which is reduced with siRNA knockdown (D) and vorinostat (B). HDAC-6 precipitates with HIF-1α in DoxR but not WT SJSA-1 cells or DoxR cells treated with Vorinostat (C).
Either direct or indirect HDAC knockdown reduces invasiveness of DoxR cells
The effect of HDACIs on invasiveness in SJSA-1 and MCF-7 DoxR cells were determined by the Matrigel in vitro invasion assay. Both invasion of SJSA-1 and MCF-7 DoxR cells were significantly reduced by pre-treatment with vorinostat (fraction of invasion 0.455 versus 0.124, P < 0.005; 0.131 versus 0.068, P < 0.037, respectively) (Figure 4A and B). Likewise, isoformspecific siRNA knockdown of HDAC-6 reduced the invasiveness of SJSA-1 and MCF-7 DoxR cells (0.455 vs. 0.056, P < 0.002; 0.131 versus 0.066, P < 0.031 respectively) (Figure 4A and B).

Figure 4. Matrigel in vitro invasion assay (mean and standard error) demonstrating histone deacetylase (HDAC) inhibition with vorinostat and small interfering RNA reduced the invasiveness of doxorubicin-resistant (DoxR) SJSA-1 (A) and MCF-7 cells (B) relative to their parental wildtype (WT) cells, (*) indication of significance (P < 0.05).
HIF-1α knockdown reduces invasiveness of DoxR cells
Using shRNA, we directly targeted HIF-1α for knockdown to confirm it was the target of the HDAC inhibitors since HDACIs regulate a wide variety of proteins and cellular functions. Western blot analysis confirmed knockdown of HIF-1α protein levels with HIF-1α-specific shRNA in doxorubicin-resistant (DoxR) SJSA-1 cells, as depicted in Figure 5A. Matrigel in vitro invasion assay demonstrated reduced invasiveness of shRNA-mediated HIF-1α knockdown DoxR cells compared to DoxR cells (0.457 vs 0.057, P <0.023) (Figure 5B and C).

Figure 5. Small hairpin RNA (shRNA)-mediated inhibition of hypoxia inducible factor-1α (HIF-1α) reduced the invasiveness of doxorubicin-resistant (DoxR) cells. Western blot demonstrated successful shRNA knockdown of HIF-1α expression (A). Small hairpin RNA-mediated knockdown of HIF-1α reduced the invasiveness of SJSA-1 DoxR cells in the Matrigel in vitro invasion assay (B), (*) indication of significance (P < 0.05). Invasion was calculated as the percentage of cells able to invade through a membrane coated with Matrigel during a 24 h period as a fraction of the control. Bars represent the normalized invasion indices (mean ± standard error). Light microscopy (2.5X) images of the Hema 3-stained Matrigel membranes demonstrated the increased number of SJSA-1 DoxR cells able to invade the Matrigel matrix compared to the parental wild-type (WT) cells (C). The drastic reduction in the invasiveness of DoxR cells with HIF-1α shRNA knockdown is apparent.
This study demonstrates that HDAC-6 plays an important role in regulating HIF-1α and may connect drug resistance and invasiveness in osteosarcoma and breast cancer cells in vitro. Drug resistance and tumor metastasis are two major properties of malignant tumors that have been studied extensively. There is evidence suggesting a relationship between the two phenotypes [1,4,25]. However, conflicting results were reported by several groups. While some studies have demonstrated enhanced invasive or metastatic ability of drug-resistant cancer cells, others have found the opposite [6,21]. Because the mechanism of the relationship between drug resistance and metastatic behavior remains unclear, the purpose of the current study was to determine the underlying mechanism(s) connecting drug resistance and metastasisassociated properties.
Our initial finding demonstrated that osteosarcoma SJSA-1 breast cancer MCF7 breast cancer MCF7 DoxR cells are more invasive than their parental WT cells, which suggests that MDR cancer cells display high invasive and metastatic behavior. In addition, metastatic cancer cells confront a different microenvironment when expanding and invading adjacent tissues. We evaluated the anchorage-independent growth of DoxR cancer cells by soft agar assay. The results showed that both SJSA-1 and MCF7 DoxR cells had a higher ability to form colonies as compared to their parental WT cells. Colony formation assay was also used to evaluate the clonogenic ability of the SJSA-1 and MCF-7 cells WT and DoxR cells under both normoxic and hypoxic conditions, our results indicate that both osteosarcoma and breast cancer DoxR cells had more formed colonies as compared to their parental WT cells. In addition, these cells may switch into a more stem-like status under hypoxic stress, and develop more aggressive phenotype when compare to normoxic condition.
Over-expression of the drug transporter P-glycoprotein (P-gp, also known as ABCB1) is a hallmark for resistance to topoisomerase inhibitors such as doxorubicin and etoposide. We have continuously found this transporter overexpressed in MCF7 and SJSA-1 DoxR cells compared to their parental WT lines (data not shown). P-glycoprotein, encoded by the mdr1 gene, has also been correlated with aggressive and invasive cancers [23,24,26]. Reduction of P-gp levels by siRNA reduced the migration of MCF7 breast cancer cells in transwell migration and Matrigel invasion assays [7]. Using similar assays, a doxorubicin-selected, MDR human melanoma line expressing high levels of P-gp showed a more invasive phenotype than the parental WT line [27]. Although it is possible that other genetic changes contributed to this phenotype, knockdown of P-gp by siRNA substantially reduced the invasiveness of this cell line in vitro. Therefore, dissection of the pathways regulating the expression of mdr1 will be beneficial in understanding the relationship between drug resistance and metastasis. Our lab has previously reported that sirtuin 1 (Sirt1), an oxidized nicotinamide adenine dinucleotide (NAD+)-dependent, class III HDAC, was over-expressed in drug-resistant neuroblastoma cells and directly controlled expression of mdr1 [28]. We have also reported that siRNA-mediated knockdown of HDAC-4 in stress-resistant cells enhanced their sensitivity to the DNA-damaging drug doxorubicin [19]. These results suggest that epigenetic modifications by HDACs might be implicated in the development of drug resistance in cancer cells. Stronach EA, et al. reported that HDAC-4 expression significantly increased in platinum-resistant ovarian tumors [29]. Our results in the current study have also showed that HDAC-6, were upregulated in DoxR cells compared to their parental WT lines. These data suggest that HDAC inhibition could provide a novel class of treatment for patients with therapeutically resistant cancers [30].
An additional gene of interest that we and others have shown to be modulated by HDAC inhibition is HIF-1α. Hypoxia-inducible factor-1α and its transcription may directly regulate the mdr1 gene [31,32]. We have also found that activity of the mdr1 promoter was significantly enhanced with cotransfection with HIF-1α (data not shown). Hypoxia-inducible factor-1α has been implicated in the radioresistance of colon cancer, cervical cancer and malignant gliomas. Recent reports demonstrated that hypoxic cells, in addition to being more chemoresistant, become resistant to apoptosis, and are more likely to migrate to less hypoxic areas of the body. Hypoxic cells accumulate stabilized HIF-1α protein and produce proangiogenic factors, such as VEGF, which stimulate formation of new blood vessels from existing vasculature, increasing tumor oxygenation and, ultimately, tumor growth. For this reason, hypoxic tumors are highly angiogenic and aggressive.
Under normoxic conditions, the HIF-1α subunit is constitutively expressed, hydroxylated and rapidly degraded. Prolyl hydroxylase domain proteins PHD1- 3 hydroxylate newly synthesized HIF-1α at prolines (P) 402 or 564, which are then recognized by the pVHL E3 ubiquitin ligase, targeting HIF-1α for proteasomal degradation [8,33]. The HIF-1α subunit is also hydroxylated at asparagine (N) 803 by FIH-1 (factor inhibiting HIF-1), which prevents HIF-1α from binding to the histone acetyltransferase p300 and inducing transcription of HIF-1 target genes. Under hypoxic conditions, newly synthesized HIF-1α is not hydroxylated and is not recognized by pVHL for ubiquitin-dependent proteasomal degradation, and therefore HIF-1 interacts with p300 to activate transcription of HIF-1 target genes. The HIF-1α protein is also regulated by O2-independent mechanisms. Heat shock protein (Hsp) 90 is a key molecular chaperone in maintaining HIF-1α stability [34]. In contrast, HIF-1α degradation is promoted by receptor for activated C kinase 1 (RACK1), Hsp70 and C terminus of HSC70-interacting protein (CHIP) [35,36].
In addition to hydroxylation, HIF-1α protein can be post-translationally modified by reversible lysine (K) acetylation, which can be pharmacologically modulated by HDACIs [14,15,37,38]. Treating cells with HDACIs reduces HIF-1α protein levels under normoxic, hypoxic and hypoxia-mimic conditions [14,37]. We have shown that HDAC inhibition reduces the level of HIF-1α in DoxR cells. Inhibition of HIF-1α is dependent on the 26S proteasomal degradation system, but can be pVHL-independent [14,37]. Two acetylation sites within HIF-1α have been identified at residues K532 and K674 [36-39]. Currently, the mechanism and functional consequences of HIF-1α acetylation/ deacetylation at different lysine residues are unclear. While the identity of the HDAC isozymes responsible for deacetylating K532 is unknown [14], K674 deacetylation is mediated by Sirt1 [40]. Qian DZ, et al. [15] and Geng H et al. [40] had shown that the inhibition of class II HDAC isozymes HDAC-4 and HDAC-6 via siRNA inhibits HIF-1α in pVHL-null kidney cancer cell lines. The HDAC-6 siRNA-mediated HIF-1α inhibition is thought to be related to acetylation of Hsp90, which disrupts its chaperone function for client proteins, including HIF-1α [15,41].
A proposed mechanism of action for chemotherapeutic agents is the generation of cytotoxic radicals via a process that depends on the presence of O2. In this study, all cells in normoxia study were incubated in a standard 5% CO2 incubator. This approach ensured that the lack of radical formation resulting from decreased O2 would not be a confounding variable in explaining the increased chemoresistance observed in cells. Deferoxamine (DFO) is a hypoxia-mimetic agent that stabilizes HIF-1α by preventing the binding of pVHL to HIF and thus O2-dependent proteasomal degradation [42]. After treatment with DFO for 16 h to prevent degradation of HIF-1α, we observed that HIF-1α levels were increased in MCF7 and SJSA-1 DoxR cells relative to their parental WT lines (Figure 4B). Inhibition of HDACs with vorinostat also reduced the level of HIF-1α protein in DoxR cells.
In non-drug-resistant cell lines, multiple studies have revealed that HDACs play a role in control of HIF-1α [15]. Fath DM, et al. demonstrated independent control of HIF-1α by HDACs in renal cell carcinoma [14]. Qian DZ, et al. did so as well, specifically showing increased HDAC-6 can increase HIF-1α expression [15,43]. In this study, we also showed that HDAC-6 and HIF-1α were expressed in DoxR cells. Moreover, inhibition of HDAC-6 either indirectly, via vorinostat, or directly, via siRNA against to HDAC-6, decrease expression of HIF-1α while decreasing the invasiveness of the cells. We also showed that knockdown of HIF-1α alone reduces invasiveness, sensitizing DoxR cells to doxorubicin. Therefore, we not only found evidence that HDAC plays a role in HIF-1α regulation in osteosarcoma, but we have done so in a drug resistant cell line.
Understanding the control of HIF-1α is important since it is implicated in so many cancer cellular processes. Although not an extensive list, it is involved in resistance to apoptosis, over-expression of drug efflux membrane pumps, vascular remodeling via VEGF, angiogenesis, metastasis, and regulation of the mdr1 gene involved in drug resistance [8,28]. By demonstrating HDAC-6 and HIF-1α expression was increased in DoxR compared to wildtype cells, that HDAC-6 inhibition decreased HIF-1α expression and invasiveness in DoxR cells, and that Knockdown of HIF-1α suppressed the aggressive behavior of DoxR cells, we have shown that HDACs indeed play a role in HIF-1α regulation and may connect drug resistance to invasiveness in osteosarcoma. Overall, these findings shed light on the implication of HDAC inhibition in drug resistance and cancer invasion. Future experiments to better understand the relationships between drug resistance and cancer metastasis will require intensive investigation.
We demonstrate that HDAC plays a significant role in regulation of HIF- 1α and may provide a link between drug resistance and cancer invasion in osteosarcoma and breast cancer cells. To broaden this work, we are currently verifying the connection with various cancer cell lines and are creating an in vivo model. Understanding the relationship between drug resistance and cancer invasion could lead to more effective drug treatments.
The following authors have no financial disclosures: F.C, N.J.S, T.B.L, S.S, M.B.M.
Fei_Chu@rush.edu; Nicholas_j_skertich@rush.edu (N.J.S.); TLautz@ luriechildrens.org (T.B.L); sszajek19@gmail.com (S.S.); MaryBeth_Madonna@ rush.edu (M.B.M)
John W. Anderson Foundation (Valparaiso, IN), the Super Jake Foundation (Gurnee, IL) and the Michael Reese Research and Education Foundation (Chicago, IL), Beckley Foundation (Chicago, IL) and Alex Manderino Foundation (St Joseph, MI).
All authors attest that they meet the current ICMJE criteria for Authorship.
None.
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
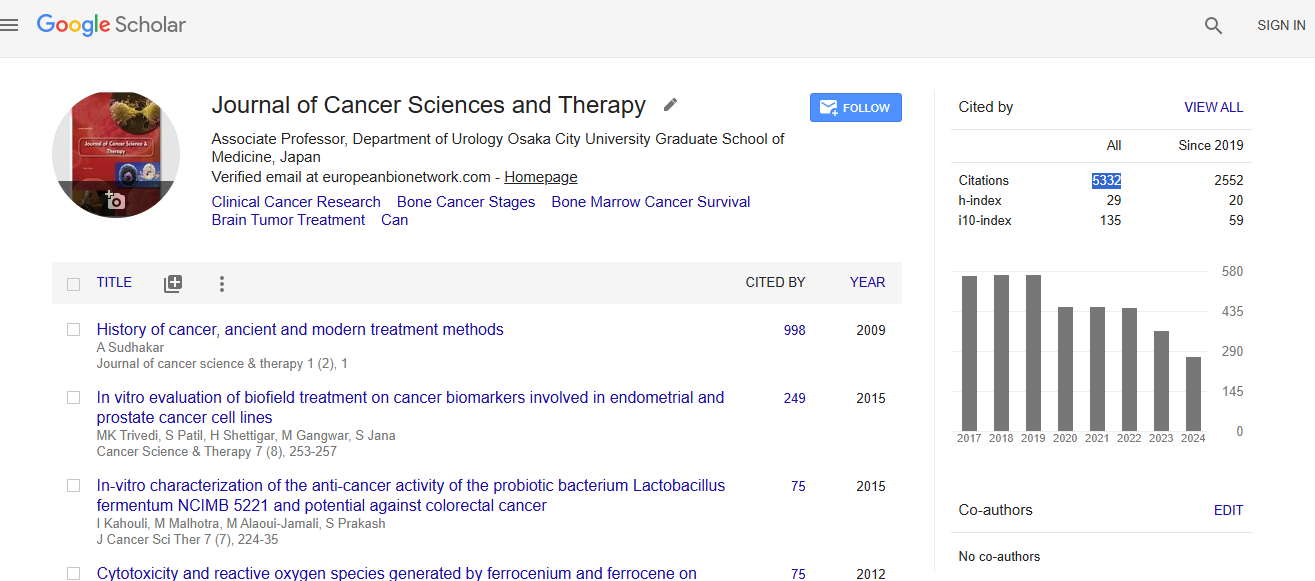
Cancer Science & Therapy received 5332 citations as per Google Scholar report