Perspective - (2022) Volume 11, Issue 9
Received: 02-Sep-2022, Manuscript No. JMMD-22-84249;
Editor assigned: 05-Sep-2022, Pre QC No. P-84249;
Reviewed: 17-Sep-2022, QC No. Q-84249;
Revised: 22-Sep-2022, Manuscript No. R-84249;
Published:
30-Sep-2022
, DOI: 10.37421/2161-0703.22.11.368
Citation: Ariel, Alyssa. “Comparative Genomics to Gene Function, Future Microbiology.” J Med Microb Diagn 11 (2022): 368.
Copyright: © 2022 Ariel A. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Microbiology is at a turning records next-technology sequencing has discovered genetic complexity amongst micro organism that would rarely were imagined through pioneers which includes Pasteur, and Koch. This facts cascade brings extensive capacity to enhance our information of character bacterial cells and the genetic foundation of phenotype variant. However, this revolution in facts technological know-how can't update set up microbiology practices, imparting the venture of a way to combine those new strategies. Contrasting comparative and purposeful genomic processes, we evoke molecular microbiology concept and set up exercise to give a conceptual framework and realistic roadmap for next-technology microbiology [1].
Experimental processes for reading micro organism have modified dramatically over moving in reaction to public hobby and fuelled through technological advances, information of those extraordinary organisms keeps to swiftly develop. We now recognize greater than ever earlier than approximately the metabolism, environmental context and host interactions of microbes, and the fee of discovery suggests little signal of slowing. Among the maximum influential shifts in generation has been the growing use of massive sequencing datasets in studies exercise. These present day studies processes keep to advantage momentum, increasing into ever greater innovative methods of the use of sequencing facts to find out complicated styles of behaviour and attain a deeper information of the bacterial mobileular. This speedy development has many conceptual advantages however has additionally come at a extensive fee, as laboratories warfare to combine those strategies and follow high-quality studies practices to new kinds of facts.
The importance and complexity of massive sequencing datasets could make them seem summary to the non-specialist, probably main to subjective decisions approximately whether or not to agree with the analyses or now no longer. This can, in turn, chance standard disenfranchisement of microbiology researchers farfar from genomic facts, selling an over-reliance on outdoor proofs or validations to present that means to sequencing-primarily based totally datasets. Here, we argue for an incorporated destiny for microbiology that mixes the strengths of conventional microbiology with the promise of emergent sequencing technology. Addressing the widening hole in studies exercise, we talk a number of the maximum influential methodologies, the validation of findings from massive sequencing datasets, and the way comparative and purposeful genomics may be incorporated to develop microbiology from essential discovery to present day microbiology studies exercise. It has been properly over a decade for the reason that nexttechnology sequencing systems have become broadly to be had for microbial genomics. The fee of sequencing has endured to fall to a degree in which massive collection datasets are withinside the price range of maximum studies agencies. This democratisation of generation become now no longer pushed through a essential alternate in how DNA is sequenced.
Here, the big numbers of character sequencing reads are mixed to put off mistakes in base calling and generate high-self belief ensemble averages. The 2nd mode is a counting feature used to survey combined populations of DNA or RNA molecules. Here, every sequencing study is tested individually, separated into agencies and scored. This method can, for example, be implemented to degree the relative frequencies of mRNA tiers in a mobileular or to seize the composition of a bacterial populace from an environmental sample. Determining the genetic foundation of phenotype variant is some of the maximum pervasive pursuits in microbiology. This is a first-rate venture and calls for information of ways adjustments to genes, and their constituent DNA sequences, can modify gene feature and have an effect on a phenotypic alternate over time. Two of the maximum transformative strategies that deal with this in micro organism are genome-huge affiliation research. Both strategies are powered through, however every makes use of exclusive capabilities of DNA sequencing technology. GWAS calls for genomes from a couple of traces inside a populace to perceive genomic factors which might be statistically related to a given phenotype or environmental circumstance and consequently makes use of the high-accuracy feature of NGS [2].
In contrast, Tense profiles fewer traces and makes use of the DNA counting feature to perceive transposon insertions in populations of mutants to perceive the contribution every gene makes to bacterial survival withinside the precise experimental context. The gene highlighted in orange represents an idealised output for every method. GWAS panels in standard, samples used for GWAS research are immediately remoted from the surroundings of hobby. If editions fall above a positive chance threshold saturated transposon libraries are grown withinside the presence and lack of the choice stress of hobby. Transposon-genome junctions from every member of the library are amplified and sequenced. The datasets received from libraries with and with out the choice stress are then in comparison to perceive the contribution of every gene to the fitness. Areas of the genome with a exclusive sample of transposon insertions are deemed to be related to the choice situations. Initially the effects of each strategies are established statistically and primary insights into gene feature are received thru literature and database searches. Deeper research affirm the genotype-phenotype dating of the effects with a purposeful validation withinside the laboratory the use of a number of experimental processes
The big software of bacterial GWAS has been made viable through adapting the methodological and analytical assumptions of human GWAS in crucial methods. First, bacterial GWAS now no longer most effective goals homologous collection variant however additionally pursuits to perceive the severe accent genetic factors and genes that can be located in some, however now no longer all, isolate genomes. Accounting for this populace shape is in particular crucial while thinking about the genetics underlying phenotype variant as cavial editions can be co-inherited with connected loci that can haven't any adaptive feature. In especially dependent bacterial populations complete clusters of traces may also proportion factors which have facilitated their enlargement in addition to people who clearly replicate not unusualplace ancestry. To deal with this, populace subsampling linear combined fashions and phylogenetic timber may be integrated into analyses to account for the clonal body of the populace. Resultant institutions that can't be defined through the impact of shared ancestry can constitute convergent genomic signatures in agencies of divergent traces. This gives clues to the evolutionary forces appearing at the bacterial genome [3].
Sophisticated bioinformatics analyses of ever large genome collections are incorporating quantitative trait variant conditioning on a couple of genomic or phenotypic determinants However, even as bacterial GWAS processes gain from maintaining the herbal populace placing of a given phenotype, they regularly go back many lots of genetic factors related to complicated trends which includes host affiliation or virulence. In such cases, it could be extraordinarily hard to perceive the position of character genes and get to the bottom of the myriad interacting selective consequences that form the located genomic variant. For this it can be vital to transport past in silico statistical institutions and recognize the feature and significance of precise genes below greater cautiously managed situations [4].
Observing how small genomic differences, in in any other case isogenic traces, have an impact on the phenotype gives proof approximately the purposeful effect of collection variant. Over decades, microbiologists have recognized the feature of severa genes throughout a couple of species especially thru investigating the impact of gene loss. It is viable to deduce gene feature through inactivating precise genes, generally thru advent of a selected mutation into the genome of an organism and evaluating the ensuing phenotype to that of a ‘wild-type’ pressure [5]. While that is exceedingly onerous in comparison to staring at genomic variant in herbal populations in silico, it gives plenty more manage of the genomic variant and the situations wherein the gene feature is being examined.
Extending the precept of gene inactivation for genome-huge purposeful research, ordered gene deletion libraries were generated for numerous version laboratory traces In those libraries, all non-vital genes were disrupted through the insertion of antibiotic markers, permitting the speedy screening of phenotypes below exclusive selective situations. While this permits investigations into bacterial species which might be tough to govern genetically, it can be hard to generate enough mutations for entire gene insurance withinside the screen, in particular because the entire genome of every pressure desires to be sequenced to find a mutation.
None.
None.
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
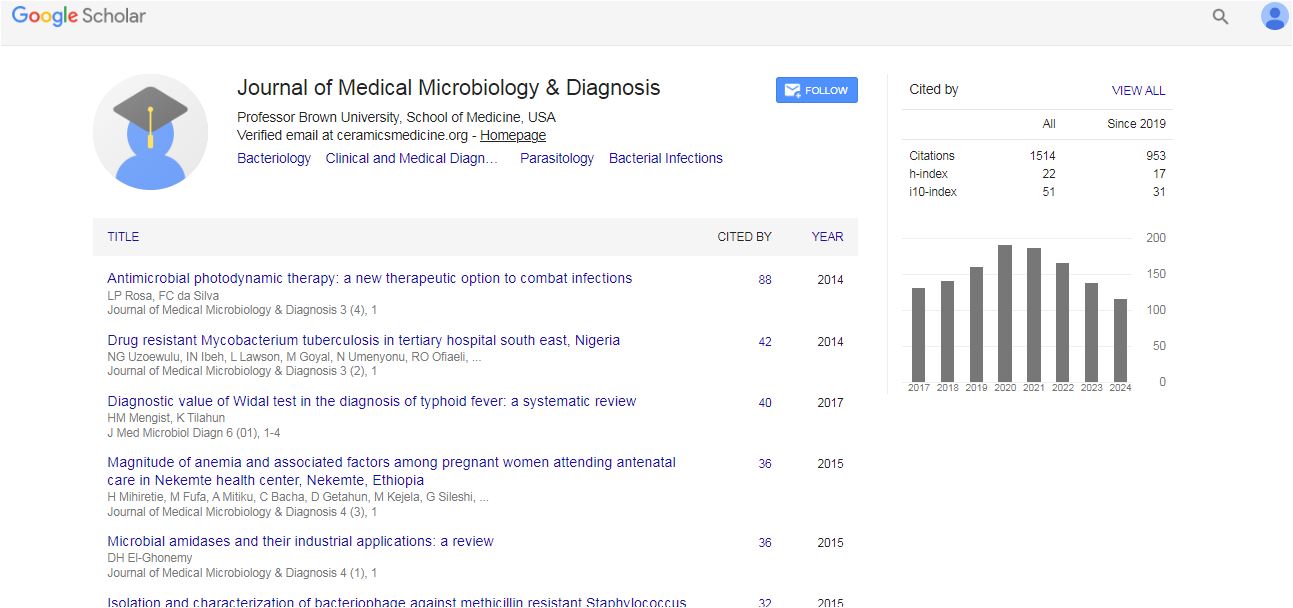
Medical Microbiology & Diagnosis received 14 citations as per Google Scholar report