Case Report - (2022) Volume 8, Issue 1
Received: 11-Jan-2022, Manuscript No. elj-22-51556;
Editor assigned: 13-Jan-2022, Pre QC No. P-51556;
Reviewed: 27-Jan-2022, QC No. Q-51556;
Revised: 02-Feb-2022, Manuscript No. R-51556;
Published:
10-Feb-2022
Citation: Etarhuni, Samira A., Abdulbast Mahdi Sharfddin, and Millad Alsaid Ghawil. “An Overview of Early Infantile Epileptic Encephalopathy 75 (EIEE75) in Libyan Boy due to Novel Mutation of PARS2 Gene Comparing with Other Reported 8 Cases around the World.” Epilepsy J 8(2022): 151. DOI: 10.37421/elj.2022.8.151
Copyright: © 2022 Etarhuni SA, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Background: Early infantile epileptic encephalopathy 75 (EIEE75) is a very rare form of epileptic encephalopathy due to mitochondrial disease, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Early infantile epileptic encephalopathy 75 is an autosomal recessive form characterized by onset of severe refractory seizures in the first months of life. The aim of the study is to compare our clinical case with previous reported cases in aspect of clinical features, MRI finding and genetic finding, and to highlights the importance of WES analysis for the diagnosis of rare disease.
Materials and Methods: The parents of our patient gave their written consent to carry out the investigations reported. One year old child product of consanguineous marriage presented at age of 5-months due to global developmental delay and epilepsy, MRI show diffuse brain atrophy. Collecting data from all reported cases due to PARS2 gene mutation leading to EIEE 75 and comparing study with our Libyan case was done.
Results: WES identified the homozygous variant c. 499G >A p(Ala167Thr) in PARS2 gene (OMIM:618437). Both parents carry the same gene in heterozygous state. As we comparing our case and the previous reported 8 cases, all are identical phenotypically, put the gene of our patient is a new mutation in homozygous state.
Conclusion: The EIEE 75 is very rare disease, there are only 8 reported cases worldwide, the disease may be under estimated, farther researches are indicated to highlights all area of it.
• Early infantile epileptic encephalopathy (EIEE) • PARS2 • Prolyl-tRNA synthetase
Epileptic Encephalopathies (EE), are a group of age-related disorders characterized by intractable epilepsy and electroencephalogram (EEG) abnormalities that may result in cognitive and motor delay [1,2]. The early infantile epileptic encephalopathy (EIEE) is heterogeneous group of neurological diseases characterized by early onset of epilepsy started from early age with progressive global developmental delay [3,4]. It is considered the most severe and earliest form of epilepsy [5]. Depending on gene mutation type there is a large group of EIEE ranging from EIEE1 to EIEE 85 [OMIM Phenotypic Series - PS308350] [6]. In addition; EIEE 75 is a very rare type of EIEE caused by gene mutation of PARS2. PARS2 is a gene, encodes the mitochondrial aminoacyl-tRNA synthetases (mt-aaRSs). Mitochondrial diseases involve the respiratory chain, which is under the double control of mitochondrial DNA (mtDNA) and nuclear DNA. The complexity of mitochondrial genetics provides one explanation for the clinical heterogeneity of mitochondrial diseases, but our understanding of the pathogenesis of this diseases are still limited [7]. Mitochondrial aminoacyl-tRNA synthetases (mtaaRSs) are a group of enzymes that play critical roles in protein biosynthesis. Mutations in mt-aaRSs are associated with various diseases. As a member of the mt-aaRS family, PARS2 encoding prolyl-tRNA synthetase-2 was recently shown to be associated with as Alpers syndrome and certain infantile-onset neurodegenerative disorders in some patients [8]. Among the early‐onset mitochondrial disorders with central nervous system involvement Alpers syndrome is one of the most common phenotypes [9].
One year old Arab Libyan child product of first degree consanguineous marriage presented at age of 5 months due to infantile spasm with hypsarrhythmia on EEG, he had moderate head control, unable to set, not following and not turned to sound. His epilepsy well controlled by vigabatrin treatment, he had feeding difficulties with poor overall growth. He is a first child in the family. On examination no dysmorphic features, poor overall growth with postnatal microcephaly, pale optic disc. Hypotonia, exaggerated reflexes both in upper and lower limbs, extensor plantar responses. His parameter at one year: his head circumference was 43 cm (far below 3rd centile, his weight was 5 kg which is also is far below 3rd centile. Both parents carry the same gene mutation in heterozygous state (Figure 1).

Figure 1. Family pedigree of Libyan family showed both parents carry c. 499G>A p(Ala167Thr) in PARS2 gene in heterozygous state while their child carry same defect in homozygous that prof autosomal recessive inheritance.
His investigation showed high serum lactate and normal liver enzymes MRI brain showed diffuse brain atrophy with hypomyelination (Figure 2).
WES
Two cc of blood of our case are collected in EDTA tube and WES was performed (Table 1).
| Classification | MAF Gnom AD [%] | Zygosity | Variant | OMIM-P (Mode of inheritance) | Gene (isoform) |
|---|---|---|---|---|---|
| Likely pathogenic | 0.000091 | Hom | C,499G >A P,(ALa167Thr) Chr1,55224336 |
618437 (AR) | PARS2 (NM-152268.3) |
WES identified the homozygous variant c.499G>A p(Ala167Thr) in PARS2 (OMIM:612036)which leads to an amino acid exchange. 16 out of 21 bioinformatics in silico program predict a pathogenic effect for this variant (Figure 3). For the best of our knowledge the variant has not been described in the literature so far (HGMD 2019.3). The variant is found in .00091% of the overall population (1 heterozygous, 0 homozygous; gnomAD). In our house database of varied ethnic and clinical background it is found in two similarly affected patients in homozygous state. considering the available information the variant is classified as likely pathogenic (bioscentia) [10].

Figure 3. Show alignment view of variant in PARS2 gene in our Libyan patient.
Genomic DNA was fragmented, and the exons of the known genes In the human genome, as well as the corresponding exon-Intron boundaries were enriched using Roche NimbleGen capture technology (SeqCap MedExome Library), amplified and sequenced simultaneously by lIIumina technology (next-generation sequencing, NGS) using an lIIumina system. The target regions were sequenced with an average coverage of BB-fold. For about 98% of the regions of Interest a 15-fold coverage, for about 96% a 20-fold coverage was obtained.
Eight reported cases (five females and three males) with EIEE 75 disease, from different countries worldwide; one Swedish boy [11-14], two Japanese [15] and two chines sisters [8], and lastly one Libyan male (our case).
All cases are sharing similar clinical features that is; seizures microcephaly and global developmental delay. The age of onset was always at infancy. Most of cases showed raised serum lactate.
The MRI brain finding are showed diffuse cerebral atrophy (all cases) and hypo myelination reported in all cases except the Swedish child [11,12]. All this finding is present in MRI brain of our case.
There were 5 cases died, the age of death ranged from 4 months to 8.5 years. One case died at age 4 months due to pneumonia [8], one died at age 2years due to cardiomyopathy [11,12] and 3 polish children died around 8 due to cardiomyopathy also (Table 2).
| Age of Death | Gene Mutation | Inheritance | EEG | MRI Brain | Clinical Feature | Onset | Gender | Nationality | Patient |
|---|---|---|---|---|---|---|---|---|---|
| 2 years due to cardiomyopathy | 612036.0001 and 612036 | heterozygous mutations in the PARS2 gene | - | Cerebral Cortical atrophy | Epilepsy, progressive microcephaly, psychomotor regression, raised serum lactate | Infancy (2,5 months) | Male | One Swedish | Sofou K, et al., [11], Sofou K, et al. [12]. |
| First died at age of 8.5yearsdue to cardiomyopathy, second at 8 years due to multi organ failure, third at? 4years due to cardiomyopathy | P364R, 612036.0003 and I80T, 612036.0004 | compound heterozygous missense mutations in the PARS2 gene | Hypsarrythmia | Cerebral Cortical atrophy, hypo myelination | Refractory epilepsy. progressive microcephaly psychomotor regression dysmorphic feature, raised serum lactate in one of them | Infancy | 2 male and 1 female | Three sibs polish | Pronicka E, et al., [13], Ciara E, et al. [14]. |
| - | (V95I, 612036.0005 and E203K, 612036.0006). |
compound heterozygous missense mutations in the PARS2gene | Sever EEG abnormality | Cerebral Cortical atrophy, hypo myelination | Epilepsy, hypotonia, microcephaly, raised serum and CSF lactate | Infancy | 2 female | Two Japanese sisters | Mizuguchi T, et al. [15] |
| One died due to pneumonia at age of 4 months | (V95I and R202G, 612036.0007). | compound heterozygous missense mutations in the PARS2 gene | Hypsarrythmia | Cerebral Cortical atrophy, delayed myelination | Epilepsy, hypotonia, microcephaly, raised serum lactate | 2 female | Two Chinese sisters | Yin X, et al. [8] | |
| - | (OMIM: 612036) | homozygous mutation of PARS gene | Hypsarrythmia | Cerebral Cortical atrophy, | Epilepsy, progressive, microcephaly, psychomotor regression, raised serum lactate | Infancy | Male | One Libyan | (our case) |
Molecular genetics
All 8 reported patients had compound heterozygous missense mutations in the PARS2 gene. The mutations, which were found by whole-exome sequencing and confirmed by Sanger sequencing, segregated with the disorder in the family.
Functional studies of the variants and studies of patient cells were not performed, but each variant was classified as pathogenic according to ACMG guidelines.
In our Libyan boy with EIEE75, WES identify homozygous mutation of PARS gene (OMIM 618437), Functional studies of variant and studies of patient cells were not performed, but this variant is classified as pathogenic according to ACMG guidelines. Both parents carry same gene mutation PARS2 gene in heterozygous state.
EIEE 75 disease is very rare disease characterized by seizures microcephaly and global developmental delay. The age of onset was always at infancy. Most of cases showed raised serum lactate.
The MRI brain findings are showed diffuse cerebral atrophy (100%) and hypo myelination reported in all cases except the Swedish child. All this findings are present in MRI brain of our Libyan case.
EIEE 75 was 5 cases died, the age of death ranged from 4 months to 8.5 years. one case died at age 4 months due to pneumonia, one died at age 2 years due to cardiomyopathy and 3 polish children died around 8 due to cardiomyopathy also, WES is very important tool for diagnosis. Our case is the first case carry the new homozygous mutation in PARS2 gene, both parents carry the same gene in heterozygous state.
The author thanks the family who agreed to participate in this study and gave permission to publish this report.
The authors report no conflict of interests.
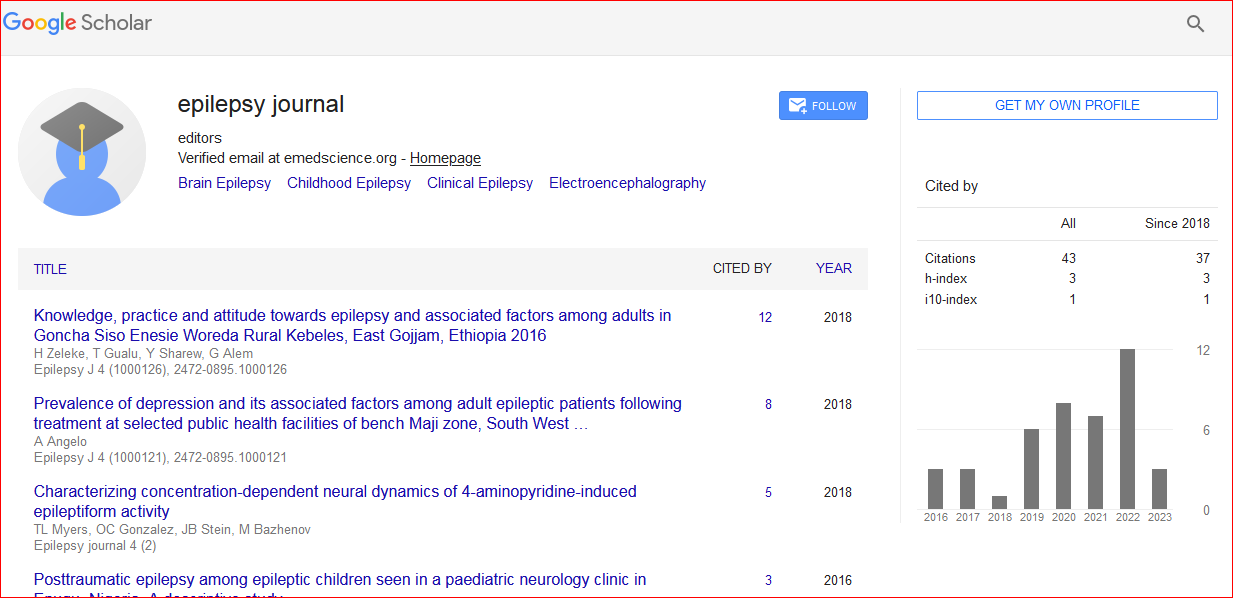
Epilepsy Journal received 41 citations as per Google Scholar report