Research Article - (2021) Volume 10, Issue 3
Received: 01-Mar-2021
Published:
23-Mar-2021
, DOI: 10.37421/2161-0703.21.10.316
Citation: Lonikar, Abhijeet, Javaria Jamil, Purnami Prashanth
and Junaid M. Khan. “17q12 Micro-Duplication.” Med Microb Diagn 10 (2021)
:316.
Copyright: © 2021 Lonikar A, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the
original author and source are credited.
Objective: We present a postnatal diagnosis of 17q12 micro-duplication with no obvious phenotypic abnormality
Case review: A female newborn weighing 2058 g was delivered NVD at 34+5 weeks of gestation to a Gravida 3 Para 2 23 year old mother, who had an antenatal history of significant maternal polyhydraminos. The baby was born flat and cyanosed with an APGAR score of 3.
Discussion: The chromosomal microarray analysis revealed a copy number gain (4 copies) of “unknown significance” of the long arm of chromosome 17 at 17q12 of size 431 kbp and encompassing 7 OMIM genes. The result also showed stretches of LOH (loss of hetrozygosity) on several chromosomes. The mitochondrial genome analysis identified a heterozygous likely pathogenic variant in the ANKRD11 gene. This result is consistent with the genetic diagnosis of autosomal dominant KBG syndrome. Physical examination revealed edema and ascites. The baby had no major dysmorphic features.
Conclusion: The 17q12 microduplication can demonstrate distinctive phenotypes, including no abnormal presentation. Nevertheless, the patient should be monitored and assessed through childhood and adulthood for any neuropsychiatric problems.
Microduplication • Chromosome • Chromosomal microarray analysis
17q12 microduplication is a rare genetic condition caused by a tiny extra part of one of the body’s 46 chromosomes-chromosome 17. Every person with 17q12 microduplication is unique and so each person will have different medical and developmental concerns. A number of common features have emerged: Children often need support with learning. The amount of support needed by each child will vary, Speech and language delay, Behavioural difficulties, Seizures, in some but not all and Otherwise generally healthy [1-7].
In families where the 17q12 microduplication has been inherited from a parent, the possibility of having another child, either a girl or a boy with the 17q12 microduplication rises to 50% in each pregnancy.
A female newborn, at 34+5 weeks of gestation weighing 2058 g was delivered by the mode of spontaneous vaginal delivery to a Gravida 3 Para 2, 23 year old mother. There is no consanguinity between the parents and no significant family history to note. The antenatal history revealed significant maternal polyhydraminos with an otherwise uneventful pregnancy. The baby was born flat with neonatal cyanosis and did not cry with Apgar scores of 3 at 1 minute and 6 at 5 minutes respectively. Neonatal resuscitation was performed and the baby was intubated. Clinical findings revealed obvious edema and abdominal distention. A chest X-ray was done which showed bilateral pleural effusion and small lung volume for which immediate aspiration was performed. Ultrasound scan confirmed ascites. Repeat chest X-ray showed tension pneumothorax and two chest tubes were inserted. The baby then improved dramatically and after 6 hours was shifted to the Neonatal Intensive Care Unit (Sheikh Shakbout Medical City).
At the time of admission, the vital signs were stable (temperature: 36.6 C, heart rate of 146 bpm, B.P-54/29, O2 saturation-99% and respiratory rate of 40 br/min) and physical examination revealed nothing more than the forementioned edema and ascites. The baby had no major dysmorphic features and a head circumference of 33.5 cm. An US Neonatal Brain Scan had done which revealed Non-immune hydrops fetalis to which the pleural effusion and ascites can be attributed. The baby was later extubated and continued to remain hemodynamically stable.
As a part of the evaluation for Non-immune hydrous fetalis, the infant underwent chromosomal microarray and mitochondrial genome analysis. The chromosomal microarray analysis revealed a copy number gain (4 copies) of “unknown significance” of the long arm of chromosome 17 at 17q12 of size 431 kbp and encompassing 7 OMIM genes. The result also showed stretches of LOH (loss of hetrozygosity) on several chromosomes. The mitochondrial genome analysis identified a heterozygous likely pathogenic variant in the ANKRD11 gene. This result is consistent with the genetic diagnosis of autosomal dominant KBG syndrome.
At 70 days of life, neurologically the baby showed no abnormalities, did not have any seizures or abnormal tone and wasn’t on any anti-eplileptic drugs. MRI Brain was done and reported normal revealing no significant findings. However, with regards to her nutrition, the baby had significant oral feeding difficulty early on and eventually needed gastrostomy. Respiratory wise, the infant is breathing comfortably without any respiratory support. There were no concerns related to her renal or cardiovascular systems with her Urea and Electrolytes within normal limits and her ECHO revealed no abnormalities.
In 46 published cases of 17q12 recurrent duplication were studied regarding the frequent features that were manifested and the study showed that the most common phenotypic features were behavioural abnormalities (94%), intellectual disability (88%), speech delay (86%), other neurologic abnormalities (85%), seizures/epilepsy (75%), dysmorphic features (71%) and hypotonia (73%). Our patient, who is 71 days of age, showed no signs of dysmorphic features, abnormal tone, seizures and abnormality of the brain on MRI. Considering the age of the infant in our case, features such as intellectual disability, speech delay, behavioural abnormalities and other neurologic abnormalities cannot be assessed at this time. A study done by showed that reciprocal duplication of 17q12 has been hypothesized to be associated with epilepsy and an increased risk of mental retardation. A study was conducted on patients with 17q12 deletion and reciprocal duplications. Patients with duplication showed cognitive impairment and behavioural abnormalities. In a review of 26 patients from 13 families with 17q12 duplication, reported by Rasmussen et al, the most consistent findings were learning disability (55%), delayed motor milestones (43%), delayed language development (43%), and a broad range of neurologic and psychiatric features. Rasmussen et al also reported 7/26 (26.9%) asymptomatic carriers with 17q12 duplication and 15/53(28.3%) asymptomatic carriers.
This could indicate that 17q12 duplication could be benign. A study from about 17q12 duplication was done and phenotypically showed a wide spectrum. Some clinical features include delayed motor milestones, wide range of neurological and psychiatric features, variable degree of learning disabilities, prolonged language development.
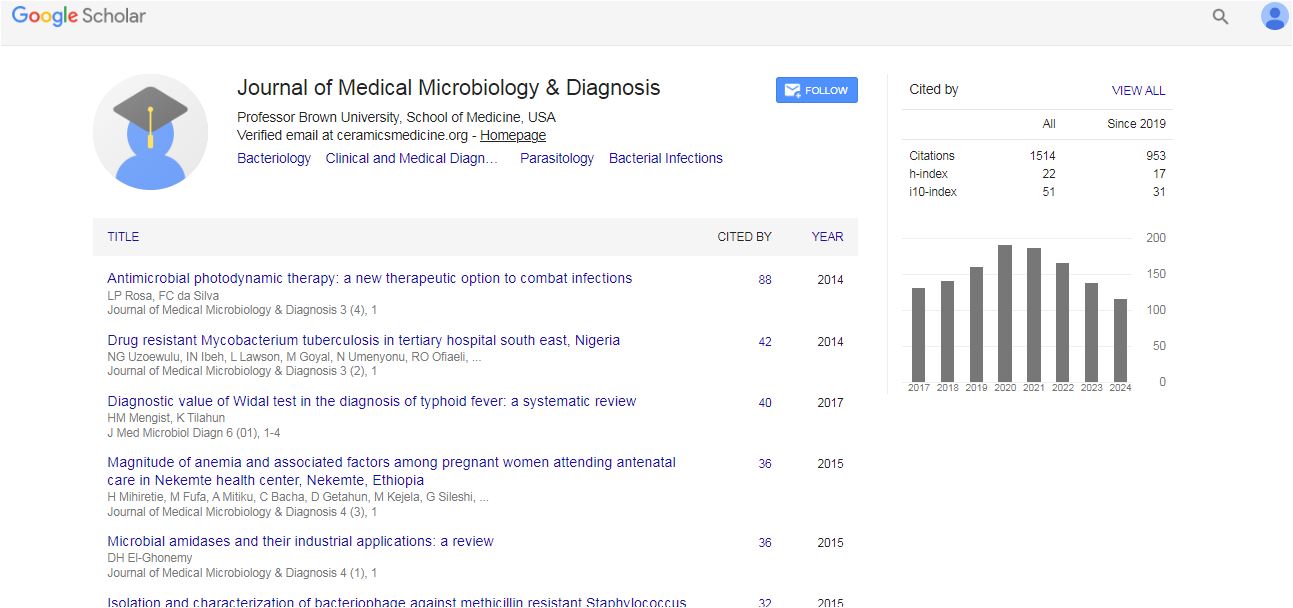
Medical Microbiology & Diagnosis received 14 citations as per Google Scholar report