Muneer Alam and Sisir Nandi
There has been a great challenge of research and discovery of novel medicinal leads against swine influenza since 2009. Rational drug design utilizing pharmacoinformatics tools has been augmented now-a-days for in-silico screening of lead compounds prior to experimental synthesis, structural elucidation, biological evaluation and finally clinical trials to make the cost efficient drug design and discovery research. There is hardly any specific chemotherapeutics for the treatment against deadly swine influenza viral infection. Therefore, it is an urgent need to design and develop new anti-viral lead compounds active against swine influenza. Quantitative structure activity relationship (QSAR) has been used to develop models that correlate biological activity of angelicin compounds derived from published literature and their computed structural properties. The approach started by generation of a series of descriptors including topological, three dimensional, constitutional, functional groups and atom fragment indices respectively solely calculated from the compounds in the data set. In this study, data set consists of 53 angelicin compounds along with their inhibitory concentration 50% (IC50, μM) against H1N1 swine influenza virus. Genetic algorithm-multiple linear regression (GA-MLR) analysis technique has been to generate a number of QSAR models. The models were validated statistically incorporating training and test set approaches. Finally, structure-based molecular docking study has been performed for interpretation of the mode of binding of the angelicin compounds toward H1N1 target. QSAR and molecular docking analysis of these congeners have not yet been reported. Therefore, this study has significant impact for designing of the highly active compounds in this series that are useful for the treatment of swine influenza. In-silico structure based docking model could be helpful for design and screening of congeneric compounds having mode of binding similarity.
PDFShare this article
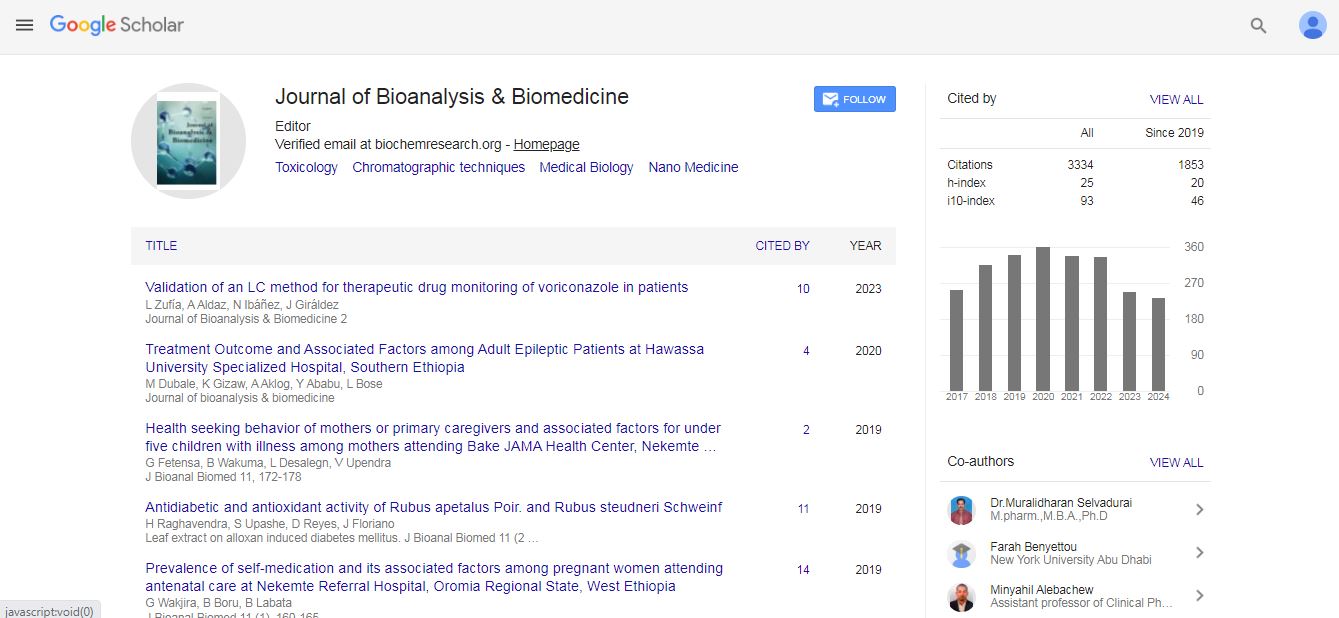
Journal of Bioanalysis & Biomedicine received 3099 citations as per Google Scholar report