Value Added Abstracts
Pages: 1 - 2Yankai Xia
The etiology and pathogenesis of Hirschsprung’s disease (HSCR) remain largely unknown. Here we employed a multiple ‘omics’-analysis to explore the important pathway related to the development of HSCR. We examined colon tissues from three independent populations with a combined analysis of metabolomics, transcriptomics and proteomics to understand HSCR. Mouse model was used for examining PGE2 induced clinical presentation of HSCR. SH-SY5Y and SK-N-BE(2) cell lines were used for examining PGE2 inhibited cell migration through EP2.The integrated analysis suggests that the level of PGE2, the expression of the genes encoding its receptor (EP2) (PTGER2) and PGE2 synthesis enzyme genes (PTGS1 and PTGES) increased in HSCR colon tissues, together with a decreased synthesis of PGE2-related byproducts. In animal study, the pregnant mice treated with PGE2 gave birth to offspring with the lack of gangliocytes in colon and gut mobility. In in vitro study, we confirmed that, when EP2 was blocked, the PGE2-inhibited migration of neural cell was recovered. Our study identified a novel pathway linking expression of PTGS1 and PTGES, level of PGE2, expression of PTGER2, and neural cell migration in HSCR, providing a novel avenue for the future diagnosis and prevention of HSCR.
Value Added Abstracts
Pages: 2 - 3Umesh Kumar
Background: Quantitative assessment of disease activity is important for effective disease management in Takayasu arteritis (TA) which is an immune-mediated inflammatory disease. The dominance of oxidative stress is the hallmark of active inflammation. Both clinical and preclinical data suggest that histidine has its strong anti-oxidative and anti-inflammatory effects [1,2]. Based on this, we hypothesized that the circulatory Histidine can serve as an indicant of active inflammation and so for monitoring disease activity in TA.
Objective: The aim of present study was to perform reliable estimation of circulatory levels of histidine and further to evaluate its potential in diagnostic screening of active and inactive TA patients.
Methods: The serum samples were collected from 98 TA patients fulfilling American College of Rheumatology (ACR) criteria and 77 normal controls (NC). The 1D 1H CPMG NMR spectra recorded on each serum sample at 800 MHz NMR spectrometer. The resulted spectra were processed and concentrations of Histidine were estimated (w.r.t formate as an internal reference) using NMR Suite of software program CHENOMX. The statistical significance was considered at p-value £0.05.
Results: According to Indian Takayasu Clinical Activity Score (ITAS) combined with acute phase reactant–erythrocyte sedimentation rate [ITAS-A (ESR)], 45 patients (46%) were clinically active, whereas 53 patients (54%) patients were inactive. Circulating levels of histidine were significantly decreased in active TA patients compared to both inactive TA patients and NC, whereas, there was no statistically significant difference between Inactive TA and NC. Further, the receiver operating characteristic (ROC) curve analysis was performed to assess the diagnostic potential of Histidine and yielded satisfactory sensitivity and specificity with AUROC equal to 0.65 [95% CI=0.54-0.76]. The circulatory levels of Histidine correlated well the erythrocyte sedimentation rate (r = - 0.19, p < 0.075) and with the C-reactive protein level (r = -0.26, p < 0.01).
Conclusion: The circulatory levels of histidine may serve as a useful biomarker for the assessment of disease activity and guiding treatment in TA patients. However, its use in clinical settings will require future studies on large patient cohorts in a longitudinal manner and procedural optimization as well to improve accuracy.
Value Added Abstracts
Pages: 3 - 5Thomas Geisberger
For the emergence of life, the origin of metabolism stands as a central problem. How did early biomolecules arise to form an intertwined network of self-controlling metabolic reactions? In scenarios for a potential chemoautotrophic origin of life, the relevant conditions could involve volcanic discharges and ashes or gradients in volcanic hydrothermal vents. These conditions can also be mimicked in laboratory settings. The analytical procedures necessary to evaluate these systems are closely related to metabolomics approaches qualified to analyze living organisms. In both cases, the main challenge is to analyze diverse mixtures of molecules in terms of quality, quantity and also isotope compositions (when working with labeled precursors). Thus, metabolomics could also provide a powerful tool to study the metabolism of “ancient” organisms for example to identify signatures of pathways which could go back to the origin of metabolism. In our work settings, methods originally developed for the analysis of bacterial pathogens [1] [2] are now transferred to organisms considered to be “ancient” or to reaction mixtures mimicking “primordial chemistry”. Analyzing extracts from these organisms or the cell-free reaction mixtures simulating origin-of-life scenarios provides a multitude of compounds. Even though many of them are still unknown, reaction mixtures mimicking conditions for the origin-of-life frequently show resemblances to extant metabolic pathways, like the formation of activated acetic acid [3], amino acids, α-hydroxacids [4], and fatty acids [5]. In turn, analysis of the “ancient” organisms is the basis for the quantitative description of hitherto unknown metabolic pathways and enzyme functions, like the dicarboxylate/4-hydroxybutyrate autotrophic carbon assimilation cycle [6] or a reverse citrate cycle involving the reverse reaction of citrate synthase [7]. In the workshop, we will present advances and examples of how methods of metabolomics have benefitted recent studies of metabolic origins.
Value Added Abstracts
Pages: 5 - 7Ruchi Baghel
In the event of an intentional or accidental release of ionizing radiation in a densely populated area, timely assessment and triage of the general population for radiation exposure is critical. Despite decades of research, counter measures still lack. In this study, we describe the potential of integrated NMR and LC-MS approaches in evaluating the radiation biomarkers. Untargeted profiling by means of broad-spectrum, highly sensitive, UPLC-ESI-QTOFMS provides a comprehensive list of metabolites at one go in a single biofluid. Present study aims to discover new, as well as validate the previously identified metabolic signatures for whole-body irradiation in mice. The study comprised 33 C57BL6 male (8-10 weeks) mice distributed as 5Gy, 7.5Gy and controls having 11 each and irradiated through60Co gamma source. Urine samples collected post24 hrswere run in both ESI positive and negative mode. All the data were normalized by sum and were than pareto-scaled followed by multivariate analysis including PCA and PLS-DA. Of the total 1514 (positive) and 1764 (negative) peaks univariate analysis(t-test, p<0.05significant) revealed a total of 658 significant (positive) molecules with creatinine (p=9.8x10-5) and L-carnitine (p=3.6x10-8)from 5Gy whereas betaine (p=2.0x10-4), 8-hydroxyquinone (p=9.6x10-5) and L-carnitine (p=3.4x10-5) from 7.5 Gy. Out of 537 significant (negative) molecules taurine (4.7x10-4) and Quinnolinic acid (6.3x10-4) were from 5Gy. Present study thus validates our previously (NMR) reported significant metabolites citric acid, hippuric acid and taurine. The results thus lay foundation for high-througput triaging by metabolomic biomarkers for effective medical management. Further pathway analysis also reveled results.
Value Added Abstracts
Pages: 7 - 8Mathias Nuamah
Background: Troglitazone (TGZ) is a member of thiazolidinedione class of chemicals was developed for the treatment of type 2 diabetes in the late 1990s. TGZ was withdrawn from the market in 2000 due to a number of fatalities due incidence of idiosyncratic liver toxicity. Till date, the mechanism of TGZ-induced liver toxicity is unclear. However, several molecular mechanisms have been proposed to underlie TGZs liver toxicity. Understanding the interactions between these mechanisms could aid drug developers more robustly predict drug-induced liver injury (DILI), a major cause of drug withdrawal.
Aims: To use a combination of in silico and in vitro approaches to examine the interaction of TGZ with multiple biological sequence causative of TGZ hepatoxicity.
Method: In silico, the Petri net software SNOOPY was used to reconstruct the known cellular effects of TGZ, including activation of PPRAy, interaction with mitochondria, activation of apoptosis. The model was imported into the MUFINS software suite and simulated. We tested the apoptotic part of our model and activation of apoptosis was validated against the published SBML model downloaded from BIOMODELS upon which the model was based. We performed In vitro assays to determine the cytotoxic effect of TGZ on liver cancer cell (Huh7 cell).
Results: The model created in SNOOPY and simulated in MUFINS was able to reproduce the behaviour for the original Biomodels submission simulated in COPASI, validating the reconstruction. Our In vitro data demonstrate that, TGZ dose dependently decreased Huh7 cell growth and viability, and induced apoptosis in Huh7 cells. Finally, we conducted caspase assays to investigate the mode of the observed cell death and did report that TGZ induced caspase 3/7 activities in a concentration dependent manner. In addition, caspase-9 activities were seen to increase in a concentration-dependent manner, however we did not record capsase-8 activities. These data support activation of apoptosis via the intrinsic route.
Conclusion: The in sillico model reproduces the behaviour of the original model and can therefore be used to explore TGZ induced apoptosis. The in vitro model system can reproduce the known effects of TGZ making it a suitable model for this current study.
Future work: Expand and parameterize the computational model predictive of TGZ toxicity to increase the predictive nature of our model. Explore TGZ metabolic pathways and conduct in vitro assays to validate our model.)
Value Added Abstracts
Pages: 8 - 9Marwa Eltoweissy
The vast majority of patients with end-stage renal disease are treated with intermittent hemodialysis as a form of renal replacement therapy. To investigate the impact of hemodialysis membrane material on vital protein removal, dialysates from 26 well-characterized hemodialysis patients were collected 5 min after beginning, during 5 h of treatment, as well as 5 min before ending of the dialysis sessions. Dialysis sessions were performed using either modiï¬ed cellulose (n=12) (low-ï¬?ux and high ï¬?ux) or synthetic Polyï¬?ux (n=14) (low-ï¬?ux and high-ï¬?ux) dialyzer. Protein removal during hemodialysis was quantiï¬ed and the dialysate proteome patterns were analyzed by 2-DE, MS and Westernblot.There was a clear correlation between the type of membrane material and the amount of protein removed. Synthetic Poly ï¬?ux membranes exhibit strong interaction with plasma proteins resulting in a signiï¬cantly higher protein loss compared to modiï¬ed cellulosic membrane. Moreover, the proteomics analysis showed that the removed proteins represented different molecular weight range and different functional groups: transport proteins, protease inhibitors, proteins with role in immune response and regulations, constructive proteins and as a part of HLA immune complex. The effect of this protein removal on hemodialysis treatment outcome should be investigated in further studies.
Value Added Abstracts
Pages: 9 - 11M M Papamichael
INTRODUCTION There has been a global surge in allergic disease with asthma control being sub-standard and challenging in children (1,2,3). Better diagnostic techniques, more effective therapeutic procedures could reduce asthma morbidity in children (4). Lipid metabolomics can be applied to identify predictive biomarkers and novel pathogenic pathways for complex chronic diseases including asthma, along with disease progression and therapeutic response (5). We investigated the correlation between plasma fatty acids (FA) and respiratory function
as biomarkers of the mild-asthma phenotype.
METHODS
Assessments:
Statistics: Spearman’s (r), P significant at 5%
RESULTS
.
CONCLUSION
Lipid metabolomics discovered new biomarkers associated adversely with pulmonary mechanics in asthmatic children. Metabolomic techniques may aid in the development of personalized prognostic, diagnostic and treatment approaches that have the potential to significantly alter pediatric asthma management.
Value Added Abstracts
Pages: 11 - 12Katsuya Nagayama
Introduction: Malignant tumors are difficult to observe in the growth process, and clarification of the phenomenon is desired. Gompertz's model (1) is said to apply to the growth of malignant tumors. Therefore, we aim to carry out numerical simulation of the growth process of malignancy and reproduce the model of Gompertz.
Method: Introduce particle model (2) as a simulation method. A particle model is a numerical analysis method that uses cell clusters as particles with physical quantities and tracks the movement of particles. In the analysis procedure, first, a blood vessel network is placed in a three-dimensional area, and cancer cell group particles are randomly generated.
Calculation conditions: Blood vessels elongate and diverge according to the amount of attractant from undernourished cancer cells nearby. The amount of attractant was inversely proportional to the amount of nutrition. For nutrient transport between blood vessels and cells, the diffusion equation is used. Cancer cells with high nutrient concentration were actively divided, and those with poor nutrition were dormant. We considered the killing by immune cells and the killing by internal pressure.
Results: Figure 1 shows the number of cancer cells grown over time, and Figure 2 shows the cancer status at the end of the calculation. In the early stage, proliferation is inhibited by the influence of immune cells. In the middle stage, some cancer cells that escaped from immune cells increased rapidly. In the late stage, the growth rate became slower because the malignant tumor became larger and the nutrient supply into the tumor worsened.
Conclusions: We performed numerical simulations from the onset of malignancy. It was confirmed that the number of cancer cells proliferated matched qualitatively to the Gompertz model.
Value Added Abstracts
Pages: 12 - 14Jinzhou Ye
Metabolite-enabled killing of antibiotic-resistant pathogens by antibiotics is an attractive strategy to manage antibiotic resistance. Our previous study demonstrated that alanine or/and glucose increased the killing efficacy of kanamycin on antibiotic-resistant bacteria, whose action is through up-regulating TCA cycle, increasing proton motive force and enhancing antibiotic uptake. Despite the fact that alanine altered several metabolic pathways, other mechanisms could be potentially involved in alanine-mediated kanamycin killing of bacteria which remains to be explored. In the present study, we adopted proteomic approach to analyze the proteome changes induced by exogenous alanine. Our results revealed that the expression of three outer membrane proteins was altered and the deletion of nagE and fadL decreased the intracellular kanamycin concentration, implying their possible roles in mediating kanamycin transport. More importantly, the integrated analysis of proteomic and metabolomic data pointed out that alanine metabolism could connect to riboflavin metabolism that provides the source for reactive oxygen species (ROS) production. Functional studies confirmed that alanine treatment together with kanamycin could promote ROS production that in turn potentiates the killing of antibiotic-resistant bacteria. Further investigation showed that alanine repressed the transcription of antioxidant-encoding genes, and alanine metabolism to riboflavin metabolism connected with riboflavin metabolism through TCA cycle, glucogenesis pathway and pentose phosphate pathway. Our results suggest a novel mechanism by which alanine facilitates kanamycin killing of antibiotic-resistant bacteria via promoting ROS production.
Value Added Abstracts
Pages: 14 - 16Sayed Metwaly
Statement of the Problem: Analysis of data is the most the most challenging step in metabolomics experiments [1]. In part, this is related to the enormous amount of data generated by metabolomics analytical methods [4]. Chemometrics, especially principal components analysis (PCA) and orthogonal partial least squares discriminant analysis (OPLS-DA) have been the most commonly used methods for analyzing metabolomics data [4]. Recently, the increase in complexity of metabolomics data sets further increased the reliance on more sophisticated supervised classification algorithms (e.g. support vector machine (SVM) and random forest (RF)) for analyzing metabolomics data. However, the lack of intuitive visualizations and the abstract nature of the output of these algorithms substantially impact their interpretability [5].
Methodology & Theoretical Orientation: Here we propose a novel algorithm, the “probabilistic universal model approximator” (PUMA). Based on PCA probabilistic mapping, PUMA projects a 3D surface that joins the points with a 50% probability of class assignment and overlays this surface on a 3D PCA scores scatterplot to delineate how the model of interest defines the interface that separates between classes. Findings: Unlike the over-optimistic OPLS-DA plots, PUMA plots are based on PCA scores scatter plots hence they impartially capture most of the data set variance irrespective of its correlation to the Y matrix (classes). PUMA’s modular design allows the examination of a myriad of classification models (such as SVM, RF, KNN, XGB, … etc.) and its interactive 3D output allows visually inspecting the performance of the model of interest (Fig. 1).
Conclusion & Significance: Careful interpretation of the output of complex machine-learning classification algorithms is becoming a necessity, given the increasing reliance on such algorithms and the abstract output they produce. PUMA is an innovative visualization approach that intuitively aid in judgement of classification algorithms’ performance and is envisaged to increase their transparency
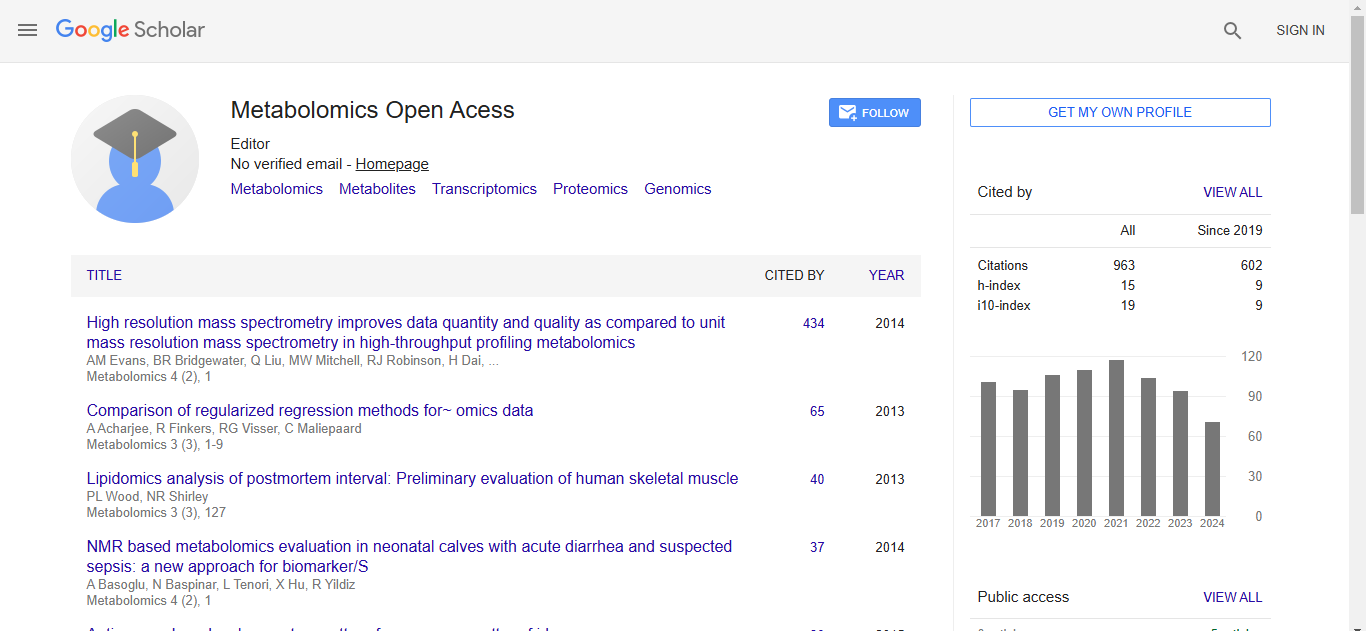
Metabolomics:Open Access received 895 citations as per Google Scholar report
 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 