Xing Li
Mayo Clinic, USA
Scientific Tracks Abstracts: J Appl Computat Math
Applying high-throughput next generation sequencing technology - RNA-seq and whole genome sequencing (WGS) - provides an unprecedented opportunity to investigate the disease-specific transcription profiles linked to potential genetic causes in Hypoplastic left heart syndrome (HLHS). Bioengineered HLHS patient-specific iPSCs and differentiated cardiac tissues offer a platform to recapitulate the individual developmental process to study the disease molecular causes. In this study, transcriptome profiling using RNA-seq was performed on iPSCs and differentiated cardiomyocytes. The results of RNA-seq data revealed over 4000 and 6000 differential genes between the family members in iPSC and differentiated cells respectively. WGS was done on blood samples of proband and parents to identify millions of variants. Variant filtering according to rarity, predicted damage and mode of inheritance pinpointed 34 genes with uncommon variants potentially involved in the pathogenesis of the disease.Ten of the 34 mutated genes displayed transcriptional differences in iPSCs while 16 of 34 mutated genes showed significantly differential expression in differentiated cells. Expression profiles for genes fulfilling both criteria (9 in total) were further characterized in iPSCs from proband and controls in a guided time-course cardiac differentiation. Two genes, ELF4 and HSPG2, displayed significantly different profiles. Of note, none of these genes had been previously linked to HLHS. In summary, by integrating the data from WGS, RNA-seq, naturally expressed time-course developmental roadmap, we triangulated a list of prioritized candidate genes that may contribute to HLHS and could be a target for future mechanistic studies for disease-specific clinical applications.
Xing Li has completed his PhD in Bioinformatics from The University of Michigan at Ann Arbor. He is an Assistant Professor in Division of Biomedical Statistics and Informatics, Department of Health Science Research at Mayo Clinic which has been recognized as the best hospital for 2014-2015 by U.S. News & World Report. He has published more than 17 papers in reputed journals and has been serving as reviewers of many journals, such as Genomics, BMC Bioinformatics, Stem Cell Research, PLOS ONE, Physiological Genomics, etc.
Email: Li.Xing@mayo.edu
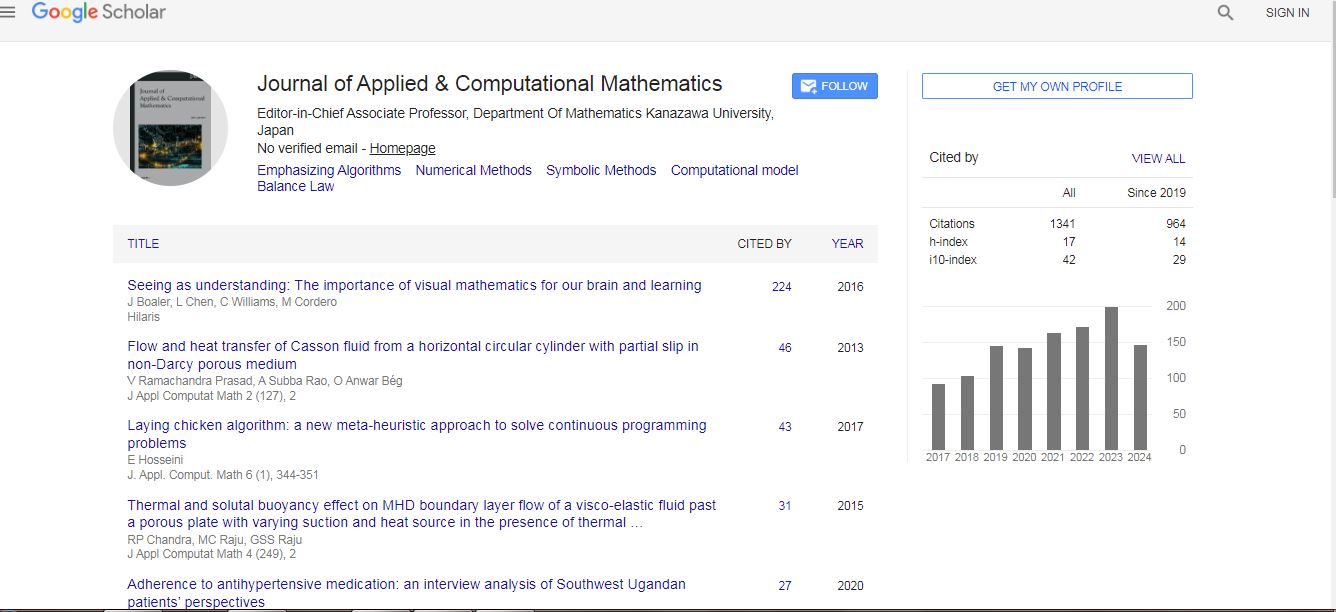
Journal of Applied & Computational Mathematics received 1282 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 