Samir Mehndiratta, Yen-Chia Huang, Fang-I Huang, Shiow-Lin Pan, Chia-Ron Yang and Jing-Ping Liou
Taipei Medical University, Taiwan
Scientific Tracks Abstracts: J Cancer Sci Ther
Histone acetylation finds its roots in the 1960s when it was recognized that acetylation of histones and remodeling of the compact chromatin structure is associated with gene induction. Histone deacetylases (HDACs) display multifaceted functions by coordinating the interaction of signal pathways with chromatin structure remodeling and the activation of nonhistone proteins; these epigenetic regulations play an important role during malignancy progression. This initiated a quest for development of inhibitors of histone deacetylase (HDAC) as novel drug candidates. We have designed and evaluated a series of potent phenyl acrylamides, based on the core structure of PXD101 and LBH589, demonstrating potent HDAC inhibition and anti-inflammatory and anti-cancer effects. The synthesized compounds are found to be endowed with potent Hela Nuclear HDAC inhibitory activity, almost 2.5 folds to 42 fold better than SAHA. Synthetics exhibited significant inhibitory effects on various cancer cell lines with GI50 values in the range of 0.02 to 0.35 μM which are 10-50 folds better than SAHA (Vorinostat). N-hydroxy-3-{4-[2-(2-methyl-1H-indol-3-yl)-ethylsulfamoyl]-phenyl}- acrylamide (MPT0G157) treatment significantly inhibited different tumor growth at submicromolar concentration and was particularly potent in human colorectal cancer (HCT116) cells. Apoptosis and inhibited HDACs activity induced by MPT0G157 was more potent than that by the marketed drugs PXD101 (Belinostat) and SAHA. In an in vivo model, MPT0G157 markedly inhibited HCT116 xenograft tumor volume and reduced matrigel-induced angiogenesis. The anti-angiogenetic effect of MPT0G157 was found to increase the hyperacetylation of heat shock protein 90 (Hsp90) and promote hypoxia-inducible factor-1α(HIF-1α) degradation followed by down-regulation of vascular endothelial growth factor (VEGF) expression. Our results demonstrate that MPT0G157 has potential as a new drug candidate for cancer therapy.
Samir is a postdoctoral research fellow in Department of Medicinal Chemistry at Taipei Medical University (TMU), Taiwan. He has his expertise in small molecules as potent anticancer compounds and his work accentuates on designing and synthesizing inhibitors of various epigenetic modulations like HDAC inhibitors, HAT Inhibitors, target based therapy, designed multiple ligand (DML) based drug design and personalized medicines for the treatment of cancer. During his doctorate, he received QS-Apple Scholarship 2014 for outstanding research and social engagements. With high impact papers in various research journals he has been awarded with Outstanding Postdoctoral Award for year 2016 and 2017 from TMU and has also received Young Research Scientist Award 2018 from SBMLS (India).
E-mail: sam131301@yahoo.co.in
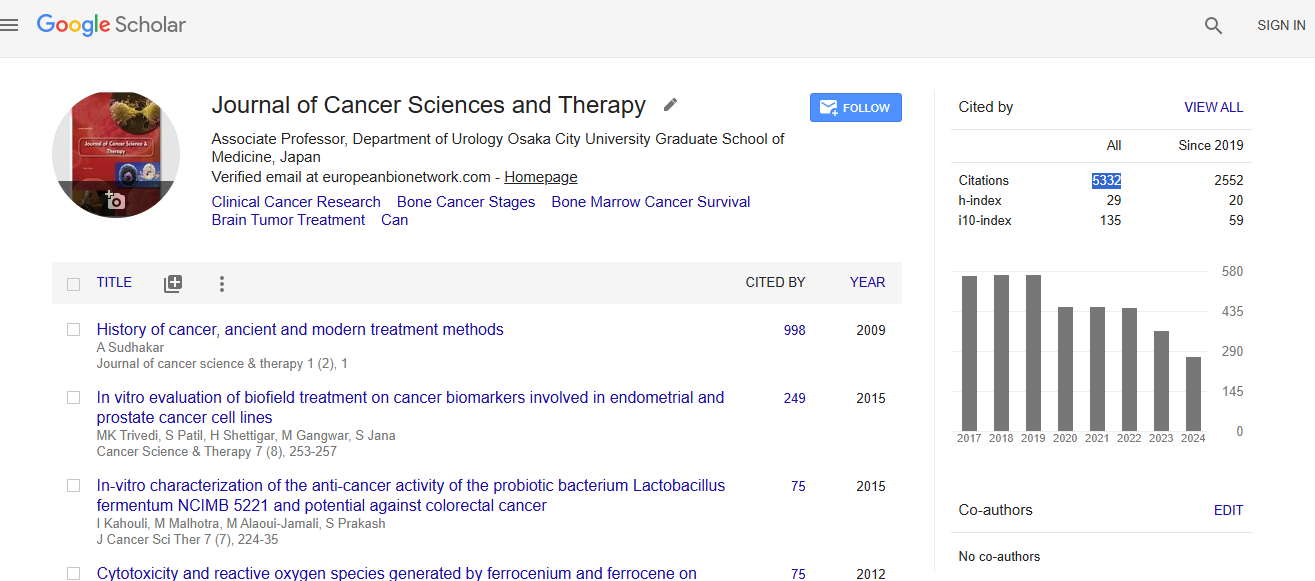
Cancer Science & Therapy received 5332 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 