Bingjie Li
University of Texas, USA
Scientific Tracks Abstracts: J Appl Computat Math
Individualized target-specific molecular medicine improves the patient managements and effective treatment of diseases in the future. The individualized medicine will emphasize the differences from both host and pathogens for every disease. The diagnosis will be determined at disease molecular levels and the treatments will be applied according to the difference of individual patient as well as pathogen. We previously demonstrated a single genetic mutation will alter the disease onset time, severity and location in the host. In this study, we intend to study the variations of a single pathogen. Clostridium difficile (C. difficile) is a gram-positive, anaerobic, spore-forming bacillus that can cause pseudomembranous colitis requiring colectomy and resulting in death in hospitalized patients. Patients receiving the first-line traditional antibiotic treatments for CDAD have initial recurrence at a rate of 25% (RCDAD). Once a recurrence occurs, there is a 45% chance of a second recurrence, and after the second recurrence, 65% of patients will have a third recurrence. We hypothesize the C. difficile has different variations and this variation could be the contributor for treatment failure. To test our hypothesis, 148 strains of C. difficile were collected from the hospital. All samples were confirmed the C. difficile presentation by culture and toxin assays. Bacterial DNA was extracted. Their molecular components were further analyzed by PCR amplification using four pairs of primers followed sequencing. Bacteria genetic characteristics were identified according to their molecular finger printing by Multilocus Sequence Typing (MLST) and dendrogram analysis. Although all bacteria are similar to the standard positive control by culture and toxin assays, biostatistical analysis reveals that they belong to different clusters by MLST. The data suggest the bacteria from environmental and infected patients are different. The bacteria in the same cluster still have genetic differences. The bacteria genetic differences may be one of the contributors that cause the treatment failure. The combination of genetic, molecular, cellular and statistical analysis will not only help us to understand the complexity of disease processes but also guide us to develop sufficient personalized disease management as well as discover more effective drugs to treat diseases.
Bingjie Li holds BM, MPH degrees and is a PhD candidate working at the University of Texas Health Science Center at Houston under supervision of Dr. Zhi-Dong Jiang. She has completed her Clinical Medicine degree from Weifang Medical University and Master of Public Health from the University of Texas School of Public Health. She is currently working on several research projects within Center for Infectious Diseases that explore pathogen variations by a combination of genetic, molecular, cellular and statistical methods.
Email: Bingjie.Li@uth.tmc.edu
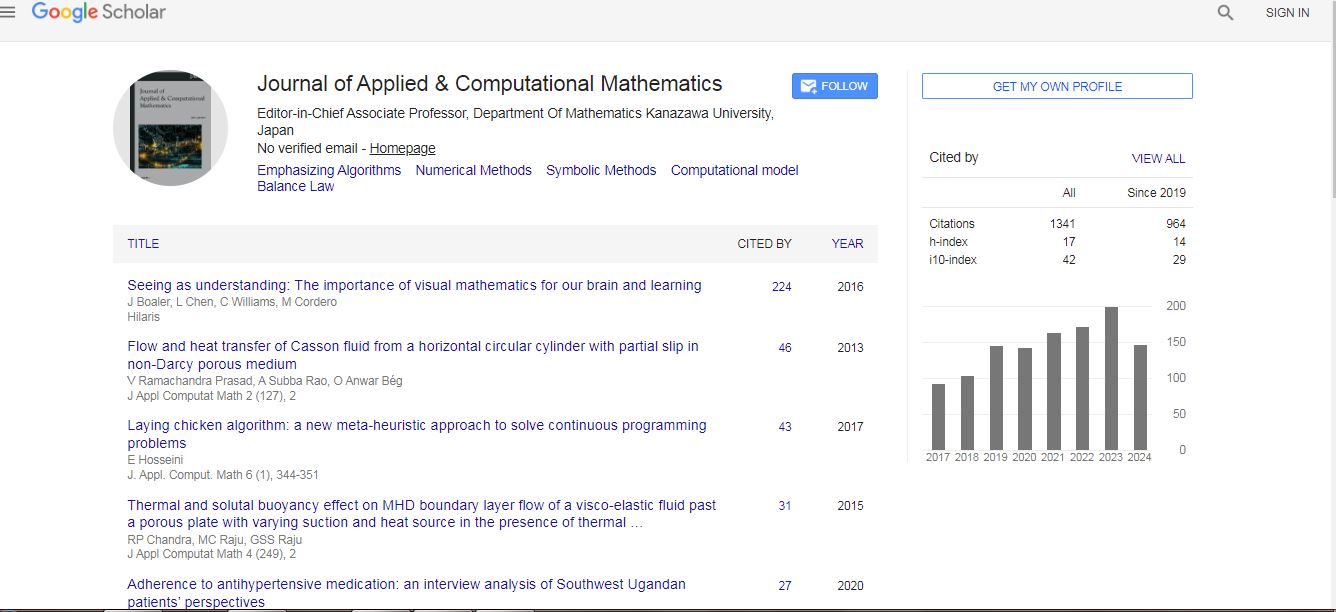
Journal of Applied & Computational Mathematics received 1282 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 