DOI: 10.37421/ 2472-100X.2022.7.181
DOI: 10.37421/ 2472-100X.2022.7.182
DOI: 10.37421/ 2472-100X.2022.7.183
DOI: 10.37421/ 2472-100X.2022.7.185
DOI: 10.37421/ 2472-100X.2022.7.184
DOI: 10.37421/2472-100X.2022.7.193
In children with craniofacial problems, upper airway blockage can be treated using a variety of surgical and maxillofacial techniques. The nasopharyngeal prong is a straightforward concept that allows the airway blockage brought on by the tongue's posterior placement as a result of a small mandible to be swiftly and easily resolved without the need for more invasive surgical treatments.
DOI: 10.37421/2472-100X.2022.7.194
DOI: 10.37421/ 2472-100X.2022.7.195
DOI: 10.37421/2472-100X.2022.7.191
Parenting a child with neurodevelopmental impairment is linked to a lower quality of life, a higher risk of anxiety and depression and greater stress (NDD). Although there is research that suggests it may be challenging to raise NDD children in LMIC, little is known about the psychological hardship these parents go through, especially in rural areas. The goal of the study was to look at the psychological distress those rural Nepalese caregivers of NDD children go through. A team of skilled mental health professionals went and spoke with 63 carers in their homes. In this study, the General Health Questionnaire was utilised to evaluate psychological distress, perceived caregiver load, disability severity and demographic information.
DOI: 10.37421/2472-100X.2022.7.192
For newborns and kids with nervous system disorders, gene-targeted medicines are now a reality. Rapid scientific progress has produced therapies that can cure or even change illness. Unresolved issues, however, include the necessity for long-term surveillance, inequities in delivery and delays and gaps in diagnosis.
DOI: 10.37421/2472-100X.2022.7.196
Indeed, even without even a trace of indications upon entering the world, kids with innate toxoplasmosis (CT) may foster serious long haul squeal, including hydrocephalus, seizures, and mental, hearing and visual debilitations, with retinochoroiditis being the most incessant sign of CT. The endanger for its improvement further down the road has been connected to the inception and timing of anti-microbial treatment after birth. In a South American populace, beginning antiparasitic treatment as soon as conceivable contrasted with a postponement until the fourth month of life or later has been accounted for to decrease the gamble of visual sores inside the initial 5 years of life from 78% to 33%. While a generally high frequency of new retinochoroidal sores during the subsequent period shows the significance of long haul follow-up for patients with CT in South America, its convenience in kids with asymptomatic disease upon entering the world has been bantered in North American and European nations.
DOI: 10.37421/2472-100X.2022.7.197
DOI: 10.37421/2472-100X.2022.7.199
DOI: 10.37421/2472-100X.2022.7.200
DOI: 10.37421/2472-100X.2022.7.198
DOI: 10.37421/2472-100X.2024.9.261
Neurosurgery plays a crucial role in the treatment of various neurological disorders affecting children. Over the past few decades, significant advancements have been made in the field of pediatric neurosurgery, resulting in improved diagnostic capabilities, minimally invasive techniques, and enhanced surgical outcomes. This article aims to explore the recent developments in neurosurgical procedures for children, highlighting pioneering techniques and their impact on patient care.
DOI: 10.37421/2472-100X.2024.9.262
DOI: 10.37421/2472-100X.2024.9.263
DOI: 10.37421/2472-100X.2024.9.264
DOI: 10.37421/2472-100X.2024.9.265
DOI: 10.37421/2472-100X.2024.9.267
DOI: 10.37421/2472-100X.2024.9.268
Pediatric oncology has witnessed significant progress over the years, leading to improved outcomes and better quality of life for children facing cancer. This article explores the advancements made in pediatric oncology, highlighting the innovative approaches and collaborative efforts that have paved the way for enhanced care for young cancer patients. Pediatric oncology focuses on the diagnosis, treatment, and management of cancer in children, adolescents, and young adults. Childhood cancers differ from adult cancers in various aspects, including tumor types, biology, and response to treatment. As a result, specialized care tailored to the unique needs of pediatric patients is essential for achieving optimal outcomes.
DOI: 10.37421/2472-100X.2024.9.269
Chromosomopathies are genetic disorders caused by abnormalities in the structure or number of chromosomes. These disorders can lead to various developmental, physical and intellectual challenges in affected individuals, often manifesting in childhood. Understanding chromosomopathies is essential for early detection, intervention and support for affected children and their families. Chromosomopathy refers to any disorder or condition caused by abnormalities in the structure or number of chromosomes. Chromosomes are thread-like structures found in the nucleus of cells, carrying genetic information in the form of genes. Any alteration in the normal structure or number of chromosomes can lead to chromosomal disorders, also known as chromosomal abnormalities or chromosomal anomalies. These abnormalities can occur during the formation of reproductive cells (sperm and egg) or during early embryonic development.
DOI: 10.37421/2472-100X.2024.9.270
DOI: 10.37421/2472-100X.2024.9.271
DOI: 10.37421/2472-100X.2024.9.320
DOI: 10.37421/2472-100X.2024.9.321
DOI: 10.37421/2472-100X.2024.9.319
DOI: 10.37421/2472-100X.2024.9.317
DOI: 10.37421/2472-100X.2024.9.316
DOI: 10.37421/2472-100X.2024.9.315
DOI: 10.37421/2472-100X.2024.9.314
DOI: 10.37421/2472-100X.2024.9.313
DOI: 10.37421/2472-100X.2024.9.312
DOI: 10.37421/2472-100X.2024.9.318
DOI: 10.37421/2472-100X.2021.6.176
DOI: 10.37421/2472-100X.2021.6.177
DOI: 10.37421/2472-100X.2021.6.178
DOI: 10.37421/2472-100X.2021.6.179
DOI: 10.37421/2472-100X.2021.6.180
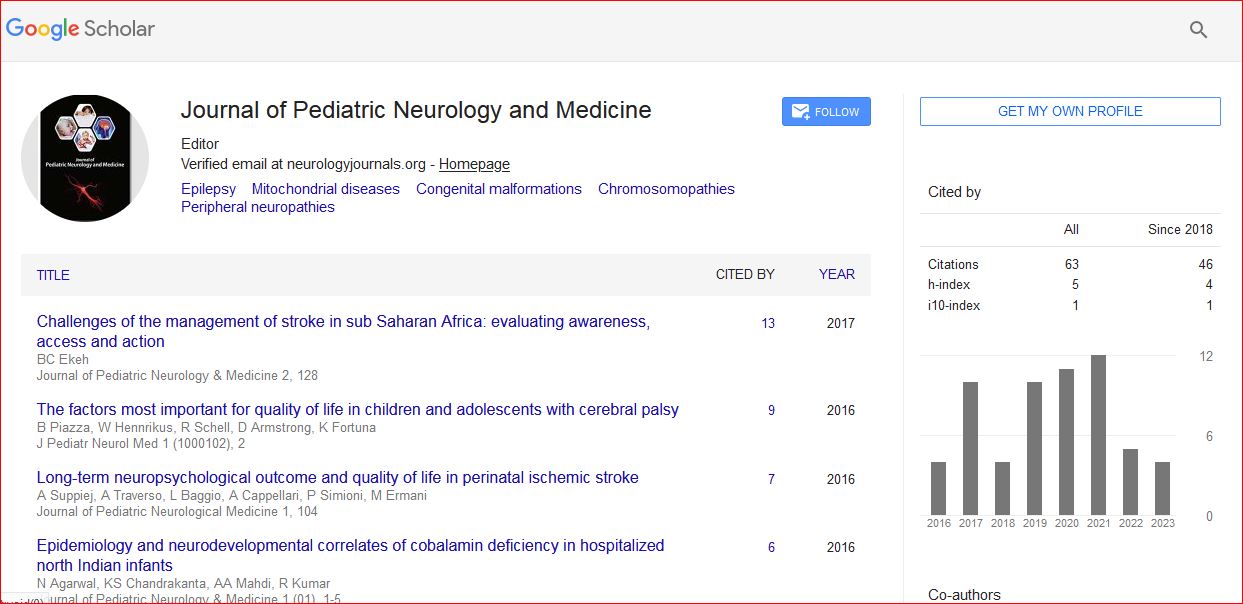
Journal of Pediatric Neurology and Medicine received 68 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 