Brief Report - (2025) Volume 10, Issue 4
Received: 01-Jul-2025, Manuscript No. jncr-26-190093;
Editor assigned: 03-Jul-2025, Pre QC No. P-190093;
Reviewed: 17-Jul-2025, QC No. Q-190093;
Revised: 22-Jul-2025, Manuscript No. R-190093;
Published:
29-Jul-2025
, DOI: 10.37421/2572-0813.2025.10.309
Citation: Santos, Miguel. ”Computational Nanoscience: Driving Innovation in Materials and Medicine.” J Nanosci Curr Res 10 (2025):309.
Copyright: © 2025 Santos M. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Computational nanoscience has emerged as a transformative field, offering unprecedented capabilities to model and understand the intricate behavior of materials at the atomic and molecular scales. This sophisticated approach is indispensable for accurately predicting and meticulously designing nanoscale properties that are paramount for a diverse array of applications, spanning critical sectors like medicine and advanced materials science. By adeptly simulating complex quantum mechanical interactions alongside larger-scale phenomena, researchers are empowered to investigate a wide spectrum of phenomena, including the nuanced dynamics of electron transport, the fascinating realm of optical properties, and the crucial catalytic activity inherent in various nanomaterials [1].
Central to the advancement of computational nanoscience is the pivotal role of Density Functional Theory (DFT), a theoretical framework that serves as a cornerstone for deciphering the electronic structures of both individual molecules and extended solid-state systems. Its application to the study of nanostructures, such as the precisely engineered quantum dots and one-dimensional nanowires, yields profound fundamental insights into their inherently unique electronic and optical characteristics. These computational revelations are instrumental in guiding the subsequent experimental synthesis and detailed characterization of these nanoscale materials [2].
Molecular dynamics (MD) simulations are equally essential for gaining a comprehensive understanding of the temporal evolution of nanoscale systems. This methodology is particularly vital for studying processes such as the spontaneous self-assembly of nanoparticles into ordered structures and for elucidating the mechanical properties of ultra-thin films. MD simulations provide invaluable dynamic information that is frequently beyond the reach of static theoretical methods, thereby offering a crucial window into time-dependent processes occurring at the nanoscale [3].
Complementing atomistic simulations, coarse-grained modeling presents a powerful and versatile approach for extending the scope of computational investigations to larger nanoscale systems and significantly longer timescales. This method strategically simplifies the representation of matter, abstracting away some atomic-level detail to enable the efficient study of emergent phenomena. It finds extensive application in areas like polymer self-assembly, the complex process of membrane fusion, and the behavior of intricate nanomaterial architectures [4].
Quantum transport simulations are absolutely critical for unraveling the complex behavior of electronic devices operating at the nanoscale. Advanced computational methods, most notably non-equilibrium Green's functions (NEGF), allow for the highly accurate calculation of electron flow through precisely defined molecular junctions and intricate nanostructures. The insights gained from these simulations are paving the way for the conceptualization and development of entirely novel electronic components with enhanced performance characteristics [5].
The profound interplay between theoretical modeling and experimental validation is a foundational principle that underpins the rapid progress within the field of nanoscience. Computational models not only serve the vital function of predicting material properties but also play an equally important role in interpreting complex experimental results. Furthermore, they actively suggest novel experimental avenues, thereby guiding the precise synthesis of materials engineered to possess specific, desired characteristics. This synergistic and collaborative approach is a powerful catalyst for accelerating both scientific discovery and technological innovation [6].
Computational methods are increasingly being leveraged for the sophisticated design and optimization of catalysts operating at the nanoscale. By meticulously simulating the intricate mechanisms of chemical reactions and the transient intermediate states involved, researchers can efficiently identify and develop highly effective catalytic materials. These advanced catalysts hold immense promise for critical applications in areas such as efficient energy conversion and robust environmental remediation strategies [7].
The optical properties exhibited by nanomaterials, exemplified by the fascinating phenomenon of plasmon resonance in metallic nanoparticles, are a subject of intense research interest. These properties are particularly relevant for a wide range of cutting-edge applications, including highly sensitive sensing platforms, advanced biomedical imaging techniques, and efficient photovoltaic devices. Computational techniques, such as time-dependent Density Functional Theory and finite-difference time-domain (FDTD) methods, are extensively employed to accurately predict and deeply understand these unique optical phenomena [8].
Computational nanoscience plays an exceptionally vital role in the burgeoning field of nanomedicine. It significantly enables the rational design of sophisticated drug delivery systems, the development of advanced imaging agents for diagnostics, and the creation of novel nanotherapeutics for targeted treatment. Computer simulations are instrumental in predicting crucial aspects like drug-nanoparticle interactions, the kinetics of drug release from carriers, and the overall biodistribution patterns of administered nanomaterials within biological systems [9].
Crucially, the development and refinement of accurate force fields represent a fundamental prerequisite for the successful application of molecular dynamics and coarse-grained simulations to complex nanoscale systems. Continuous research efforts are dedicated to the creation of force fields that are not only more robust and reliable but also more transferable across a broader spectrum of nanomaterials and diverse biological systems, thereby expanding their utility and impact [10].
Computational nanoscience fundamentally enables the modeling and comprehension of material behavior at atomic and molecular levels, proving vital for predicting and designing nanoscale properties essential for medicine and materials science. Through simulating quantum mechanical and larger-scale phenomena, researchers explore electron transport, optical properties, and catalytic activity of nanomaterials [1].
Density Functional Theory (DFT) stands as a cornerstone of computational nanoscience, facilitating the study of electronic structures in molecules and solids. Its application to nanostructures like quantum dots and nanowires illuminates their unique electronic and optical properties, guiding experimental efforts in synthesis and characterization [2].
Molecular dynamics simulations are indispensable for understanding the time-dependent behavior of nanoscale systems, including nanoparticle self-assembly and thin film mechanics. These simulations offer dynamic insights often unavailable through static methods, revealing time-dependent nanoscale processes [3].
Coarse-grained modeling provides a powerful strategy for simulating larger nanoscale systems and extended timescales compared to atomistic simulations. By simplifying matter representation, it allows investigation of phenomena such as polymer self-assembly, membrane fusion, and complex nanomaterial architectures [4].
Quantum transport simulations are paramount for understanding nanoscale electronic devices. Techniques like non-equilibrium Green's functions (NEGF) accurately calculate electron flow through molecular junctions and nanostructures, paving the way for innovative electronic components [5].
The synergy between theory and experiment is foundational in nanoscience. Computational models not only predict properties but also aid in interpreting experimental data, proposing new experiments, and guiding material synthesis toward desired characteristics, accelerating innovation [6].
Computational methods are increasingly employed in the design and optimization of nanoscale catalysts. Simulating reaction mechanisms and intermediates helps identify efficient catalytic materials for energy conversion and environmental remediation applications [7].
The optical properties of nanomaterials, such as plasmon resonance in metallic nanoparticles, are crucial for applications in sensing, imaging, and photovoltaics. Computational methods like time-dependent DFT and FDTD are used to predict and understand these phenomena [8].
Computational nanoscience is vital for nanomedicine development, facilitating the design of drug delivery systems, imaging agents, and nanotherapeutics. Simulations predict drug-nanoparticle interactions, drug release kinetics, and nanomaterial biodistribution [9].
The development of accurate force fields is critical for molecular dynamics and coarse-grained simulations of complex nanoscale systems. Research continues to focus on creating more robust and transferable force fields for diverse nanomaterials and biological systems [10].
Computational nanoscience leverages advanced simulation techniques to model and understand materials at the atomic and molecular levels, driving innovation in medicine and materials science. Key methods include Density Functional Theory for electronic structure analysis, molecular dynamics for temporal evolution studies, and coarse-grained modeling for larger-scale systems. Quantum transport simulations are essential for nanoscale electronics, while the interplay between theory and experiment accelerates discovery. Computational approaches are also crucial for designing catalysts and understanding optical properties of nanomaterials. Furthermore, these methods are vital in nanomedicine for designing drug delivery systems and therapeutics. Ongoing advancements in force field development are critical for enhancing the accuracy and applicability of these simulations across various nanoscale systems.
None
None
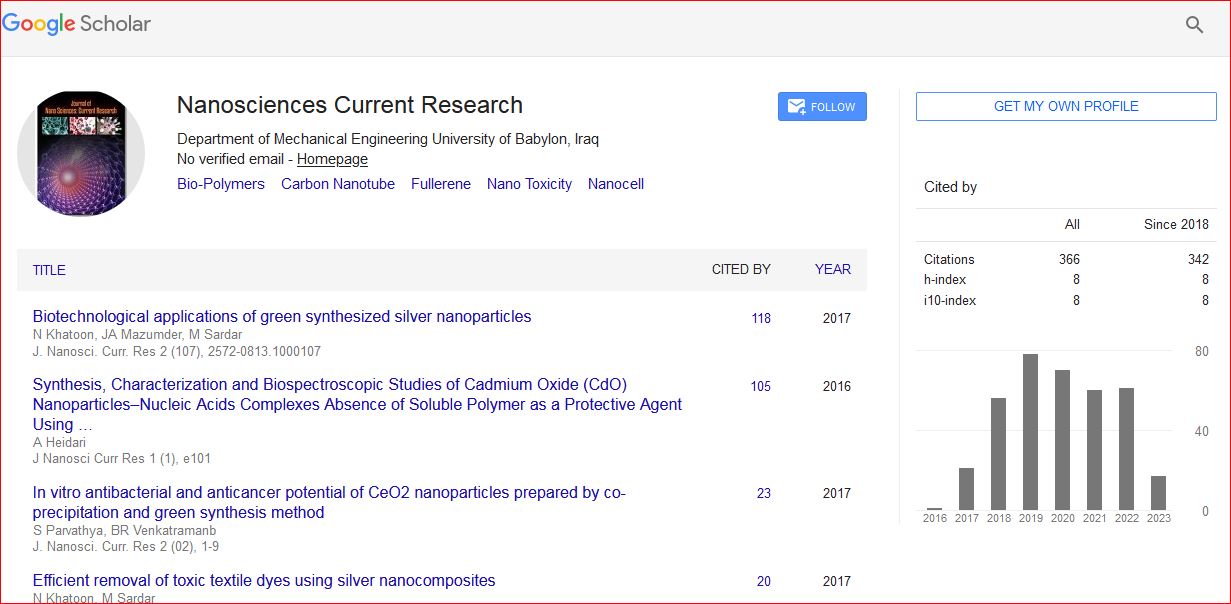
Journal of Nanosciences: Current Research received 387 citations as per Google Scholar report