In 1925 Mendes da Costa coined the term "Erythro and Keratodermia variabilis in a mother and daughter" to describe this inherited disorder which is characterized by fixed hyperkeratotic plaques and erythematous lesions with "contours such as the dividing lines sides". [3] EKV includes subtypes such as EKV Mendes da Costa and EKV Cram-Mevorah. EKV Mendes da Costa represents two thirds of all types of EKV. It is due to germline mutations in the gap junction protein (GJ) beta 3 and beta 4 which code for the connection of the proteins GJ 31 (Cx31) and 30.3 (Cx30.3). Mutations in these genes modify the structure and function of GJ channels, alter the cytoplasmic traffic of mutant GJ proteins to the cell membrane and induce cell death, thereby altering normal epidermal differentiation. [4]
EKV manifests itself at birth or during infancy. The EKV Mendes da Costa presents two types of morphological skin lesions: migratory erythematous type and fixed hyperkeratotic plaques. Initially, they come in the form of an erythema, then become hyperkeratotic. Migratory lesions can occur all over the body, persist for hours or even days, and disappear spontaneously to reappear. They have irregular borders with a fine graduation. Variations in lesions can be influenced by emotions, physical factors such as temperature, friction, pressure, or hormones. The fixed hyperkeratotic plaques are erythematous, well defined and distributed over the face, buttocks and extremities. [5] They can be associated with palmoplantar keratoderma. EKV Cram-Mevorah is a rare variant, characterized by circulating repens or erythematous gyratum such as skin lesions. They also present a fast migrating figured erythema in an annular, garland or spiral pattern. [6]
Roset erythrokeratoderma or Degos syndrome is a rare and atypical variant of EKV. It was described by Degos in 1947. It is characterized by intermittent annular lesions with central exfoliative scaling and surrounding erythema, giving the appearance of targetoid, or "in roundels" distributed over the extremities. [7] In addition to the characteristic lesions, fixed erythematous plaques and hyperkeratotic plaques may also be present. All forms of EKV persist until adulthood, but erythematous lesions are recurrent or disappear after puberty.
The diagnosis of EKV is based on clinical features and genetic analysis. Histopathology results are not specific and include acanthosis, hyperkeratosis, papillomatosis and the normal granular layer. The differential diagnosis includes PSEK, congenital ichthyosis and Netherton syndrome. The atypical variants must be differentiated from subacute lupus erythematosus, annular centrifugal erythema and multiform erythema. [8] PSEK lesions are non-migratory, well delimited, polycyclic, hyperkeratotic and distributed symmetrically on the elbows, knees, dorsal surface of the hands, feet and buttocks, and generally sparing the trunk.
2021 Conference Announcement: Metabolomics:Open Access
2021 Conference Announcement: Metabolomics:Open Access
Review Article: Metabolomics:Open Access
Review Article: Metabolomics:Open Access
Research Article: Metabolomics:Open Access
Research Article: Metabolomics:Open Access
Research Article: Metabolomics:Open Access
Research Article: Metabolomics:Open Access
Editorial: Metabolomics:Open Access
Editorial: Metabolomics:Open Access
Editorial: Metabolomics:Open Access
Editorial: Metabolomics:Open Access
Scientific Tracks Abstracts: Molecular and Genetic Medicine
Scientific Tracks Abstracts: Molecular and Genetic Medicine
Scientific Tracks Abstracts: Journal of Tissue Science and Engineering
Scientific Tracks Abstracts: Journal of Tissue Science and Engineering
Posters & Accepted Abstracts: Journal of Tissue Science and Engineering
Posters & Accepted Abstracts: Journal of Tissue Science and Engineering
Scientific Tracks Abstracts: Journal of Tissue Science and Engineering
Scientific Tracks Abstracts: Journal of Tissue Science and Engineering
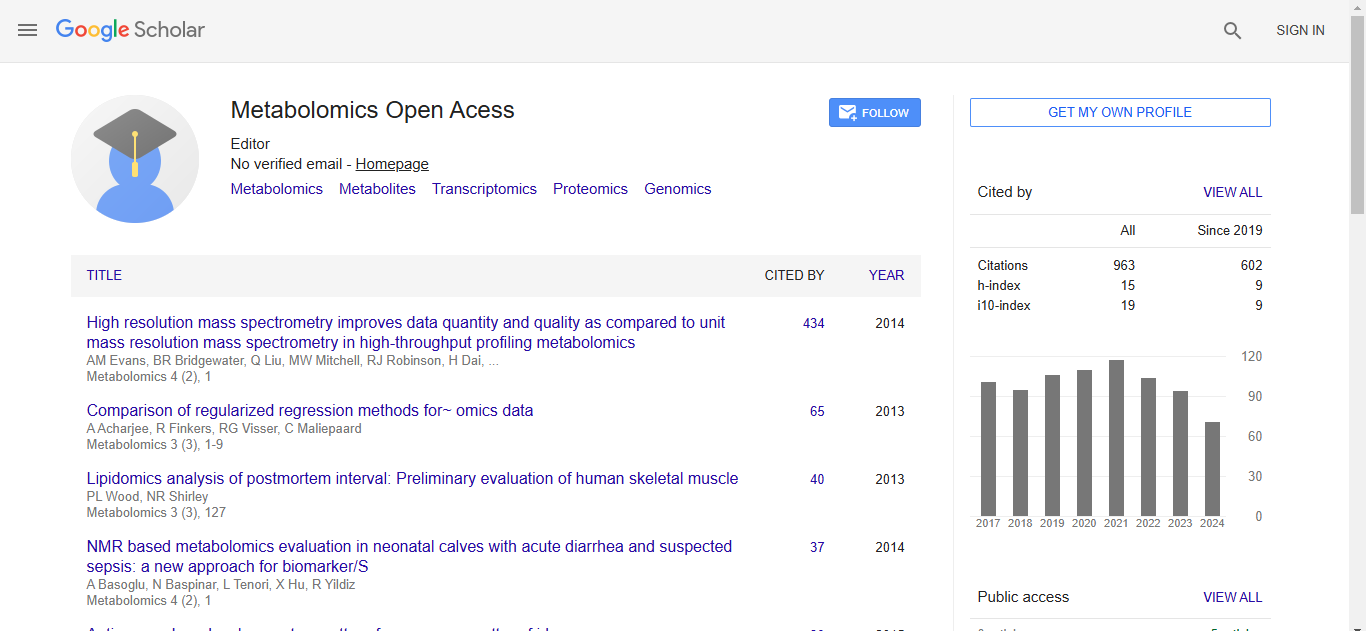
Metabolomics:Open Access received 895 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 