Kyung Hee Do
Ubaldo Armato
Abbreviations: Aβ: Amyloid β peptide; Ach: Acetylcholine; AD: Alzheimer’s Disease; BDNF: Brain-Derived Neurotrophic Factor; BFCSNs: Basal Forebrain Cholinergic Septal Neurons; DGy: Dentate Gyrus; GN: Granule Neuron; LTP: Long-Term Potentiation; NGF: Nerve Growth Factor; NT-3: Neuro-Trophin-3; p75NTR: p75 NeuroTrophin Receptor; SGZ: Sub-Granular Zone; SST: Somatostatin; SSTR3: SST Receptor 3; SVZ: Sub-Ventricular Zone; TA Transit-Amplifying; tPA: tissue Plasminogen Activator.
The key role of p75NTR in the generation of DGy GNs. In this cheme, a neuro-trophin (BDNF or NGF) in the DGy SGZ activates cilial p75NTR, the signals from which stimulate progenitor cell proliferation and the generation of rapidly proliferating TA precursor neurons. After several rounds of proliferation, the TA precursor neurons stop proliferating and signals 6 from the cilial SSTR3 and BDNF•TrkB complexes stimulate them to mature, enter the granule cell layer and serve transiently as intensive long-term potentiation (LTP)- generating, super-sensitive encoders of novel multi-modal inputs flowing along the perforant pathway from the entorhinal cortex. Normally, half or more of the newborn cells may die without reaching the granule cell layer. But NGF produced in the DGy from proNGF by tPA-generated plasmin forms NGF•Trk A complexes that stimulate BFCSNs to make Ach and release it in the hippocampus, which promotes newborn neurons survival and thus neurogenesis and memory formation. Eventually, the successful neurons lose their hyper-responsiveness and become loaded with old ‘memories’, retire, and are replaced with new recorders.
The Aβ peptides accumulating in the early stages of AD also stimulate cilial p75NTR like the neuro-trophins and with this the proliferation of granule cell progenitors and their TA progeny. But the additional neurogenesis expected from this stimulation fails and neurogenesis drops because proNGF can no longer be converted into mature NGF because of a developing shortage of plasmin resulting from an Aβ1-42-induced fall in tPA. But proNGF preferentially binds and activates the p75NTR•sortilin complexes instead of NGF•TrkA complexes on the BFCSN axons. The retrograde flow of p75NTR•sortilin signals down the axons kills the BFCSNs and stops the hippocampal supply of ACh that would otherwise promote the survival of the TA precursor neurons. Furthermore, TA neurons that do survive are prevented from further maturing by the lack of SST?a hallmark of the AD brain?and the consequent silencing of SSTR3 signaling.
More than 90% of the murine DGy GNs have a ~4-µm-long cilium protruding from them. These cilia are loaded with p75NTR, the signals from which can stimulate granule cell progenitor proliferation, and also with SSTR3 (not shown), the signals from which can drive the postmitotic maturation of newborn neurons.
So what might the cilially restricted p75NTR do for adult neurogenesis? Key clues to its function are: (i) proliferating (i.e., BRDU-positive) cells in the DGy SGZ express p75NTR; and (ii) knocking out p75NTR reduces the proliferating cells and hippocampal neurogenesis by 59%-79% [12,13]. Although we do not yet know whether the progenitor cells in the other adult neurogenesis region, the SVZ [1], also confine p75NTR to their primary cilia, BDNF-, NGF- or Aβ 1-42-induced p75NTR signaling stimulates their proliferation and neurogenesis without requiring BDNF's or NGF's corresponding TrkB or TrkA co-receptors. Therefore, cilial p75NTR is a driver of the proliferative stage of adult neurogenesis.
The ability of the proliferogenic cilial p75NTR to bind and be activated by Aβ 1-42 can explain a so far mysterious aspect of adult neurogenesis in early AD. Progenitor cell proliferation and adult neurogenesis normally decline with age, and it would be expected to at least continue dropping with the approach of AD. But the ability of Aβ 1-42 to activate the proliferogenic p75NTR reverses this trend and progenitor cell proliferation counter-intuitively increases in the early Aβ 1-42-accumulating stages of AD. While this is happening, the pro-NGF-activated axonal p75NTR.sortilin complexes induced by the accumulating Aβ 1-42 start killing BFCSNs and cutting off the hippocampal supply of ACh. Also counter-intuitively despite the increased Aβ 1-42/p75NTR-driven progenitor cell proliferation, neurogenesis is not increased because fewer TA neurons can survive without the support of ACh and because the cilial SSTR3 receptors needed for maturation and memory functions are silenced by the lack of SST in AD brains.
In conclusion: Cilial p75NTR must now be part of models of adult neurogenesis in the DGy and memory formation and of the cognitive decline in aging and AD brains.
Note: This work is partly presented at 5th Global Experts Meeting on Parkinsons, Huntingtons & Movement Disorders, Oct 30-31, 2019 Tokyo, Japan
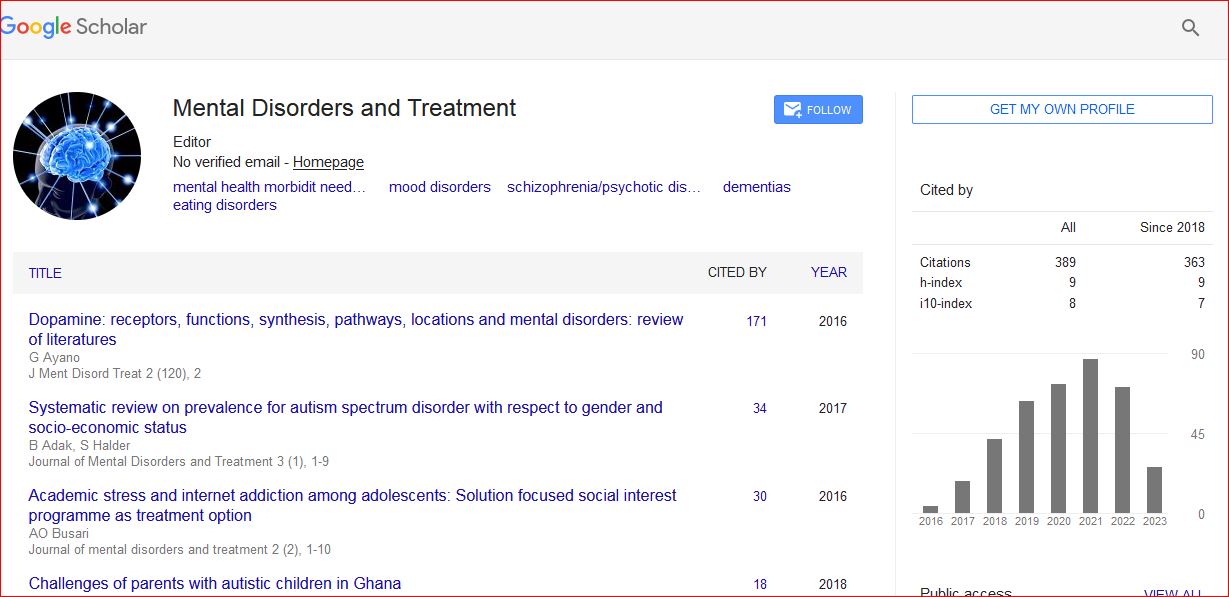
Mental Disorders and Treatment received 556 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 