
Ivan P Gorlov
Department of Community and Family Medicine
Dartmouth College, Hanover, NH, USA
The overarching goal of my research is to address challenges resulting from common use of technologies generating high throughput data. Next generation sequencing platforms routinely produce millions of data reads. As a result, multiple statistical tests are required making it difficult to sort signal from noise. We can however turn those and other challenges into advantages. In many cases multiple data types are available for the same phenotype. Integrating data across various platforms may enhance and consolidate true signal allowing its easier discrimination from noise. As an example, we have combined data from genome-wide association studies (GWAS) with whole-genome profiling of gene expression to identify and validate novel cancer genes (Gorlov et. al, 2009).
One of the most effective ways to integrate diverse data is meta-analysis. I have used meta-analysis of the whole-genome expression data to identify novel prostate cancer genes. We have used a modified approach based on the analysis of individual P-values. The advantage of the approach is that it can be applied to any type of data and therefore can be used to integrate different types of the data
My recent research interest was in molecular mechanisms of prostate cancer. We have conducted a meta-analysis of gene expression data from multiple studies (Int J Cancer, 2009; BMC Cancer, 2010). We also performed in silico functional profiling of individual tumors. We found that functional profiles (differentially expressed biological functions) correlate with clinical traits stronger than gene expression profiles do. I am also working on prioritizing of sequence variants identified through next generation sequencing. It is important to have paired sequence data on germline mutations (host genotype) and tumor samples from the same patient. Genotype of the host provides information on initial genetic makeup of the tumor. We can deduce evolutionary history of the tumor including the rate of accumulation of somatic mutations, clonal heterogeneity, and clonal selection by comparing host and tumor genotypes. We are also developing approaches for in-depth analysis and interpretation of the gene expression data. An important problem here is tumor heterogeneity. Tumor heterogeneity has two aspects: clonal or cellular heterogeneity within an individual tumor and heterogeneity between different tumors: different tumors may use different genes to acquire the same phenotype, e. g., drug resistance. In a recent study we found that an analysis of interindividual variation in expression levels helps to identify genes underlying tumor heterogeneity. I see my role in bridging a gap between clinical researchers and the vast publicly available data on gene expression, somatic mutations, genotyping, methylation and sequencing as well as bioinformatics and statistical tools to deal with them. Those resource help put the results of an individual study in the context of what is already known in the area. The combination of in-house and publically available data may help with prioritizing of the findings, facilitates the development of a working hypothesis and guides future experiments.
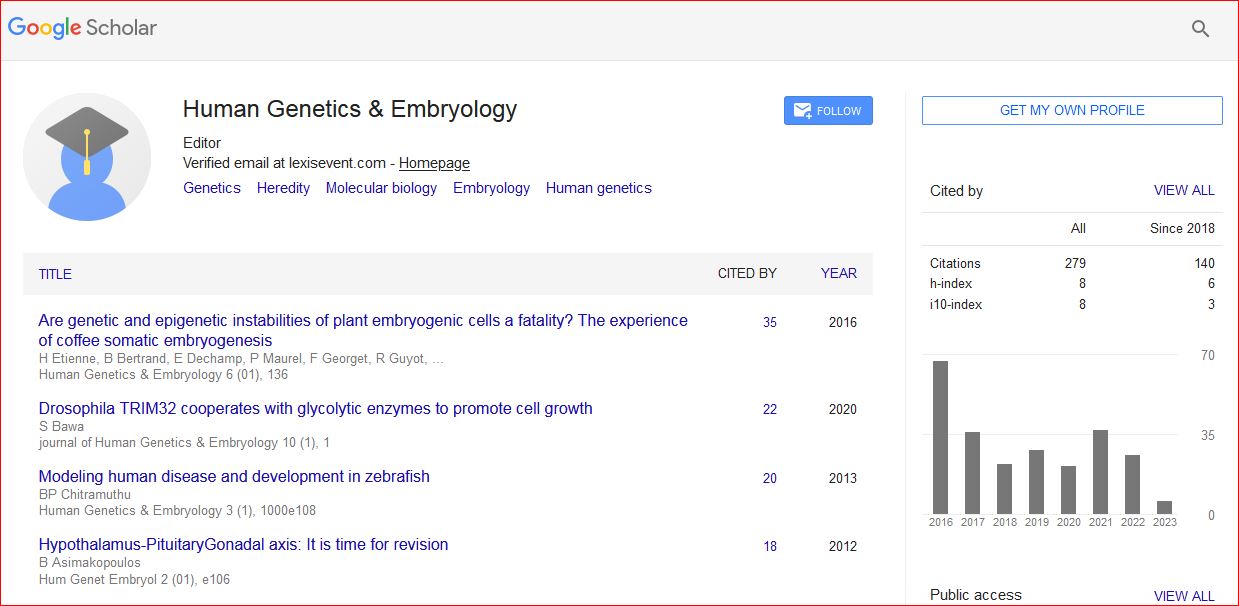
Human Genetics & Embryology received 309 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 