John K Healy
Investigation of PHAII has led to a marked expansion of knowledge of distal renal tubular electrolyte handling, which controls critical final urinary composition. This disease, found from infancy to late adulthood, is characterized by hyperkalemia, with up to 9 mmol/l plasma K+, despite a normal glomerular filtration rate. The hyperkalemis leads to acidosis, sometimes severe enough to limit growth. Hypercalciuria and renal calculi may occur. Hypertension occurs in 75% of patients. Plasma renin is depressed and plasma aldosterone is low normal. Even if normotensive at presentation, hypertension generally develops over about 30 years. The hypertension is linked to increased Na+ and Cl- reabsorption through the Na+-Cl-co-transporter (NCC) in the distal convoluted tubule (DCT). Although frequently familial, 36% of cases of PHAII are de novo. The hyperkalemia has been ascribed in humans to (a) suppression of renal outer medullary K+ secretion (ROMK), or (b) increased chloride (Cl-) reabsorption (a Cl- shunt) depressing the degree of negativity at the site of K+ secretion in exchange for Na+ at the epithelial Na+ channels (ENaC), or (c) decreased Na+ delivery to the ENaC in the late distal nephron reducing K= secretion at that site. In 2000, geneticists discovered a serine-threonine kinase in rats they called WNK1, because it lacked lysine at the usual catalytic point (with no lysine (=K)), and soon WNK’s 2,3 and 4 were found. In man, mutations of WNK’s 1 and 4 (WNK’s 1 and 4) localised to the distal nephron in the critical site of electrolyte homeostasis, and were found to cause PHAII by failing to cause suppression of the NCC, which is the function of normal WNK’s. It was eventually found that mutations in WNK1 and WNK4 cause 13% of PHAII. Recently a further system regulating distal electrolyte handling called cullin 3 and kelch-like3 was found in an E3 ubiquitination ligase complex in the distal nephron, and mutations of these (CUL3 and KLHL3) cause 79% of PHAII. Clinical severity of PHAII is, in decreasing order, CUL3, recessive KLHL3, dominant KLHL3, WNK4, andWNK1. CUL3 and KLHL3 block ubiquitination (removal for degradation) of WNK4, accumulation of which causes suppression of ROMK and so can cause hyperkalemia as in mechanism (a) above. Mutant WNK4 also stimulates Cl- reabsorption by the paracellular pathway, supportive of (b) above. Although over-activity of the NCC may initially decrease Na+ delivery to the ENaC as in (c) above, in a stable state Na+ delivery to the ENaC would be normal, and sodium sulphate and sodium bicarbonate infusion studies, with ample delivery of Na+ to the ENaC, failed to provide even a quantitatively normal kaliuretic response, weakening the argument for (c) above. Thiazides are the most effective treatment of PHAII, as they inhibit the NCC. They produce mild hypovolemia in regular use, which may cause antidiuretic hormone release and this is a potent stimulator of K+ secretion. Thus, dDAVP rapidly reverses the hyperkalemia of PHAII, and this has been used successfully in treatment of this condition. Na+ restriction should be used with caution as it has been shown to increase plasma K+ in some studies, while in others it has reduced it or had no effect. If still needed, dietary K+ restriction may be effective.
Reza Heidari
Objective: Pre-exposure of rats to normobaric hyperoxia (O2 ≥95%) may induce late pre-conditioning against renal ischemiareperfusion (IR) injury. In this study we investigated probable mechanisms of IR injury such as the role of reactive oxygen species (ROS), renal antioxidant agents, and heat shock proteins (HSP) 32 and 70 during delayed hyperoxia-pre-conditioning (HO). Methods: Fifty-two rats were divided into 7 groups: (A) IR, (B) HO + IR, (C) mercaptopropionyl glycine (MPG) + HO + IR, (D) MPG + IR, (E) HO + sham, (F) MPG + sham, and (G) sham. Rats in the following study groups (group B, C and E) were kept in a normobaric hyperoxic environment for 4 h/day for 6 consecutive days, after which they were subjected to 40 minutes of ischemia; animals in the control group (group A, D, F, and G) were kept in a normoxic cage. At the end of the preconditioning period, 24 hours of reperfusion was performed. Renal function was assessed by measuring serum creatinine (Cr), blood urea nitrogen (BUN), and creatinine clearance (CLCr). Induction of the antioxidant system was evaluated by measuring renal catalase (CAT) and superoxide dismutase (SOD) activities and glutathione (GSH) and malondialdehyde (MDA) content. The role of ROS was investigated by use of MPG (a ROS scavenger). HSP32 & 70 mRNA and protein also were determined. Results: The hyperoxia-preconditioned IR group (B) had a lower plasma Cr and BUN and greater CLCr compared with the IR group (A) (P≤0.016). Administration of MPG led to an increase in plasma Cr and BUN and a decrease in CLCr in group C compared with the hyperoxia-preconditioned group B (P≤0.004). The hyperoxia-preconditioned IR group had a greater CAT activity and GSH level compared with the IR group A (P≤0.007), whereas the administration of MPG did not change the GSH level but led to a decrease in CAT activity in group D compared with group B (P<0.001). SOD activity did not change in hyperoxia-preconditioned ischemic rats compared with ischemic rats. Hyperoxia preconditioning and MPG administration in ischemic animals did not result in any considerable change in MDA level compared with the IR group A. Also, there were no clinically relevant differences in HSP32 & 70 mRNA and protein between all groups. Conclusion: The present study demonstrates that repeated pre-exposure to hyperoxia can decrease subsequent renal IR damage in this rat model of renal ischemia. Free radical production after hyperoxia appears to play a pivotal role in the hyperoxia-induced renal protection independent of HSP level. Antioxidant enzyme activities and especially catalase seem to be implicated in this renal protective mechanism
Mohamed Shehata
Lung transplantation is indicated for terminal stage lung diseases such as chronic obstructive pulmonary disease, idiopathic pulmonary fibrosis, cystic fibrosis, pulmonary hypertension, and sarcoidosis, which have a significant impact on the pulmonary vasculature and may affect the function of the right ventricle and cardiac output. Accordingly, right ventricular and correspondingly the left ventricular parameters and functions have been reported to improve after lung transplantation. However, improved cardiac index, echocardiography and other cardiovascular parameters are not the only indicators of good prognosis. Renal functions may, in addition, provide more reliable clinical prognostic evaluation. When the cardiac output is improved, following lung transplantation, renal perfusion and the urine output would correspondingly improve. However, if renal injury develops, urine output would not be able to reflect the improvement of the cardiac functions, and the cardiovascular system may instead be affected secondary to the renal injury. High rates of incidence of acute and chronic kidney injuries have been reported following lung transplantation; with complete recovery from the acute kidney injury did not decrease the risk for the development of chronic kidney disease or long term mortality. Though renal injury following lung transplantation depends on many risk factors, including the original status of the patient’s kidneys and the effects of the immuno-suppression, especially calcineurin inhibitor therapy, the increased production of inflammatory cytokines due to the ischemic reperfusion injury and the donor-recipient contact can be propagated to significant levels that lead to renal and other organs injury, dysfunction and or failure. In addition, reducing the proinflammatory stimuli associated with lung transplantation, may affect the long term immunosuppression regime. Hence, effective EVLP might, to some degree, affect the risk of acute and orchronic kidney injuries following lung transplantation. Taking these concepts into consideration, a nonrandomized retrospective study has been recently reported by the Toronto team to compare 52 standard lung transplants to 13 EVLP transplants regarding the incidence of acute kidney injury following transplantation. The results showed no significant differences.
Sara Querido
With the introduction of combination antiretroviral therapy (cART), prognosis of HIV infection has been improved and kidney transplantation (KT) in HIV positive patients became possible. We reviewed the demographic, clinical, laboratorial and therapeutic data of all the HIV-infected patients who underwent KT prior between 2009 (first KT in Portugal in a HIV-infected patient) and May 2014. Case accrual was through all Portuguese KT centers where a KT in a HIV-infected patient was performed. Patients were transplanted following the American and Spanish guideline recommendations that included maintenance on cART, undetectable plasma HIV RNA copies and absolute CD4 counts of ≥200 cells/μl in the last 6 months. Fourteen KT were performed on men, 3 KT on women. The mean age of patients at the time of transplantation was 49.9±11.7 years. HIV status was known for 12±5 years. Eight patients had AIDS in the past and all patients received grafts from deceased donors. Twelve patients (64.7%) received induction therapy with basiliximab and two patients had early graft loss. In 2 patients humoral rejection was diagnosed and in 3 patients, cellular rejection. Two patients died and one additional patient had early graft loss. KT is a possible but challenging, renal replacement therapy in selected HIV patients. Even in those with AIDS criteria in the past, when the disease is controlled and after the reconstitution of the immune system with cART, KT can be performed. Nevertheless, the risk-benefit ratio for each patient needs to be taken in consideration.
Rizo-Topete Lilia MarÃa
Introduction: The AKI appears in 5-25% of patients in ICU, of which 6% will require RRT. If the AKI is associated with MODS mortality will be 50% and if RRT is required this will be 80%. Sepsis and Acute tubular perfusion are causes of AKI. The CRRT is an option for hemodynamically unstable patients and those who cannot handle the volume or metabolic disorders. The hemodialysis (HD) in critical patients is a common practice; however, the use of continuous therapy with hemodiafiltration modality requires special characteristics. Objective: To describe the experience using PRISMA monitor in our center. Material & Methods: Retrospective, descriptive, observational study. All patients were given CRRT with PRISMA at our center from March 2013 to November 2014. Data analysis was performed using Excel and SPSS programs. There is no conflict of interest and was conducted according to the ethics committee of our hospital. Results: CRRT was applied in an active way to 18 patients, 15 males (83%) and 3 females (17%), the average age was 43.9 years (Min. 17 Max. 78). 14 presented AKIN III, 4 where known with CKD. The most common cause of AKI was septic shock (83.3%). The oliguric AKI was the most common form of presentation in 86% of the patients. The average days of stay in ICU was 17.5 (SD 16.5). The average days of arrival and development of AKI is 2.6 days (SD 2.9). APACHE II and SOFA admission average was 30.5 (SD 6.5) and 13.6 (of 3.9) respectively. It was possible to stop CRRT in 5 of 18 patients (27.7 %), 2 patients continued with HD. There was a patient with combined therapy PRISMAMARS. Only 3 out of 18 patients (20%) survived the hospital stay. In the comparative analysis of the groups: Survivors versus non survivors, there were no statistically significant differences in the SOFA and APACHE II scores or in the days of stay in the ICU with IC of 95%. As for the prescription, blood flow measured in ml/min, extraction measured in ml/hr, the dialysate, the reinjection and total UF, showed no statistically significant differences with IC of 95%. Discussion & Conclusions: According to the results, our experience is similar to that reported in the literature with high mortality in patients with AKI and MODS, despite improvement in renal function. With the methodology used and the present number of patients, it´s not possible to point out a good or bad prediction factor on the clinical characteristics of the patients or the therapeutic prescription.
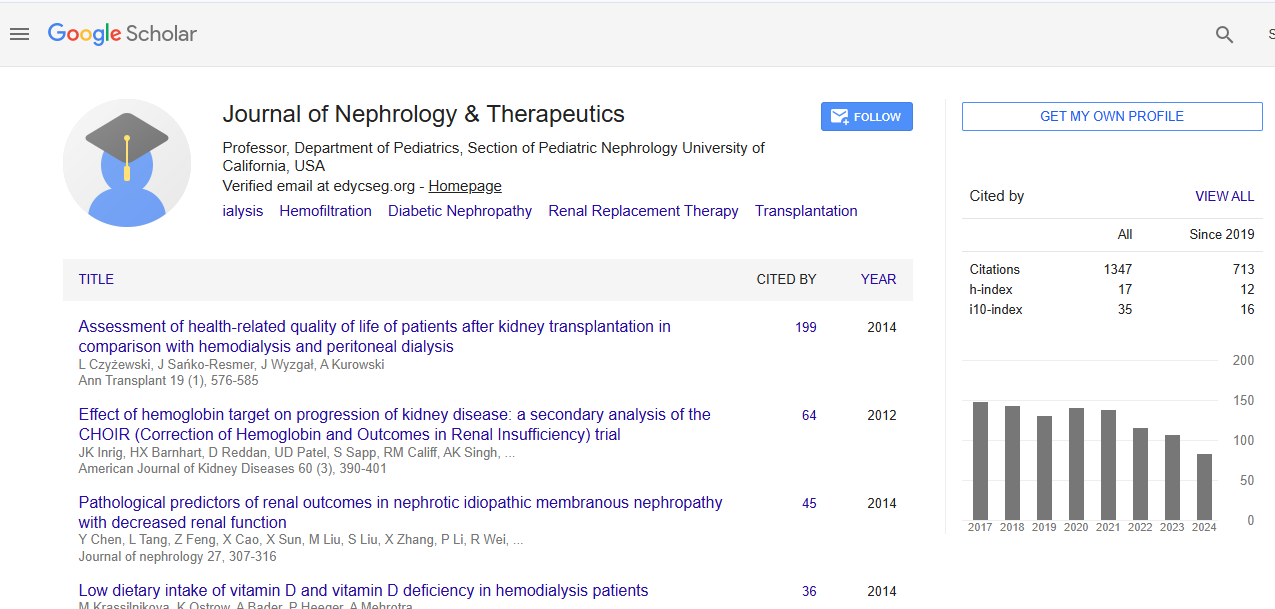
Journal of Nephrology & Therapeutics received 784 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 