Jayaraj P B
Conventional drug discovery methods rely primarily invitro experiments conducted with a target molecule and a very large set of small molecules to choose a right ligand. With the exploration space for the right ligand being very large, this approach is highly time consuming and requires high capital for facilitation. Virtual screening, a computational technique used for evaluating a large group of molecules to identify lead molecules, can be used for this purpose to speed up the drug discovery process. Ligand based drug design works by building a conceptual model of the target protein. Ligand based virtual screening uses this model to evaluate and separate active molecules for a target protein. A classes of algorithm in machine leaning called Classification algorithm can be used to build the above model. In this abstract, 3 different machine learning approaches to solve virtual screening is described. The first method utilises an efficient virtual screening technique using Random Forest (RF) classifier. Second technique applies SVM classifier for virtual screening. The third method demonstrates the applicability of Self Organizing Map (SOM) as a classifier for screening ligand molecules, which is first of its kind in this area as per the literature. The talk end with comparing the plus and minus of the three techniques. The GPU parallelisation of these methods will be also explained in details.
PDFShare this article
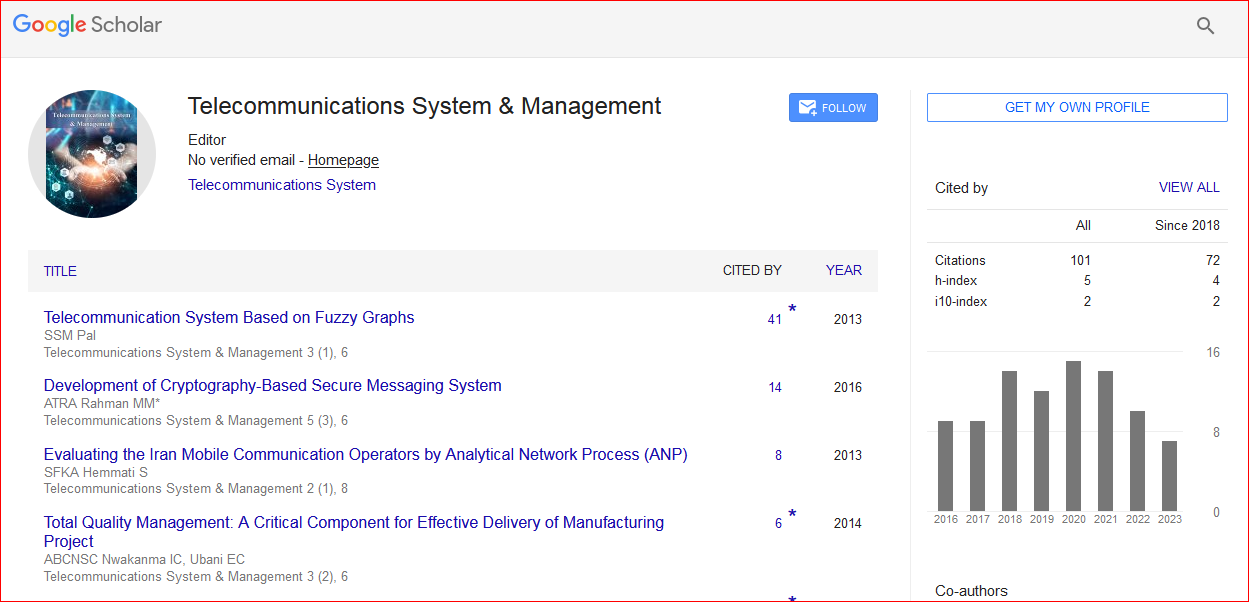
Telecommunications System & Management received 109 citations as per Google Scholar report