We present a series of cases involving individuals with the homozygous MCPggaac haplotype, a genetic configuration associated with an increased likelihood and severity of atypical hemolytic uremic syndrome (aHUS), particularly when combined with other high-risk aHUS mutations. Complement blockade therapy was administered at a median age of 92 months (with an interquartile range of 36 to 252 months). Prior to initiating CBT (Eculizumab), patients experienced a median of two disease relapses. These relapses transpired within an average span of 22.16 months (median of 17.5, ranging from a minimum of 8 months to a maximum of 48 months) following the initial subsequent disease onset (observed in 6 out of 8 patients). Treatment encompassed plasmapheresis/intravenous plasma exchange (PI/PEX), occasionally supplemented by renal replacement therapy (RRT). Upon the implementation of complement blockade, the occurrence of disease relapses ceased in the pediatric population. For children possessing the MCPggaac haplotype, with or without additional genetic mutations, achieving remission was feasible through renal replacement therapy, without an immediate imperative for complement blockade. However, in cases where aHUS relapse manifested shortly after disease onset or when relapses transpired recurrently, sustained complement blockade emerged as a necessary course of action. The specific duration of such blockade, however, remains uncertain. Failure to initiate complement inhibition prior to experiencing 4–5 relapses could potentially lead to the development of proteinuria and chronic renal failure over time.
HTML PDFShare this article
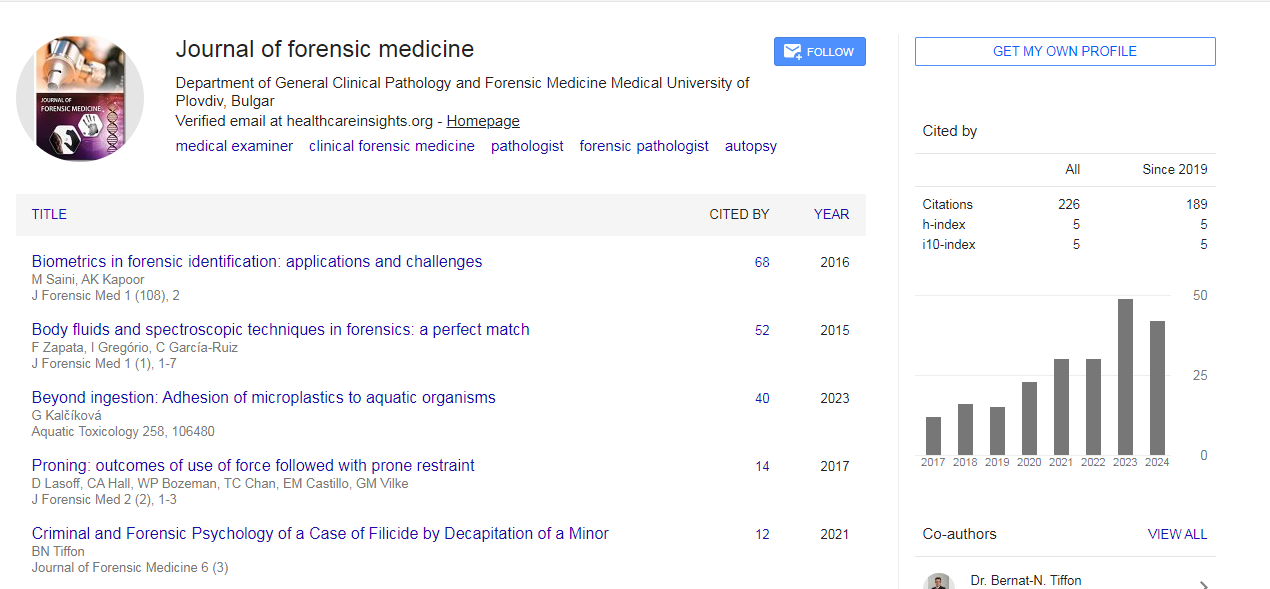
Journal of Forensic Medicine received 165 citations as per Google Scholar report