Weiwen Zhang
Microbial syntrophic metabolism has been well accepted because the heart of how methanogenic and other anaerobic microbial communities function. Dissimilatory sulfate-reducing prokaryotes (SRB) are a various group of anaerobic bacteria that are widespread in nature that play an important role within the global cycling of carbon and sulfur1,2,3. The SRB are known to possess two major lifestyles: sulfidogenic and syntrophic metabolism. In the presence of sulfate, SRB use it as the terminal electron acceptor during the oxidation of various products of primary fermentations and oxidize them to CO22,4. When sulfate is depleted, SRB in general ferment organic acids and alcohols, producing hydrogen, acetate and carbon dioxide and rely on hydrogen- and acetate-scavenging methanogens to convert these compounds to methane5,6. This working relationship of SRB with methanogens is understood as ‘syntrophy’ and has been considered because the heart of how methanogenic and other anaerobic microbial communities function7. A model syntrophic interaction involves lactate oxidation by Desulfovibrio vulgaris to supply acetate, CO2 and H2 as products, which the methanogen, Methanosarcina barkeri, can then further convert to CH48. The removal of H2 by M. barkeri provides a thermodynamically favorable condition (i.e., low H2 concentration) for the continued oxidation of lactate by D. vulgaris3,8. The advantage of having two different metabolic capabilities is that it raises the chances of survival of SRB in environments where electron acceptors could become depleted3. While the physiology of the symbiotic relationship of SRB and methanogens has been studied for several decades years3,9, relatively little is known about the genes and their expression dynamics associated with the syntrophic interactions, partially thanks to the shortage of suitable methodologies for measurements of biological properties in mixed-culture systems within the past. To address the difficulty , a transcriptomic analysis approach, which is in a position to differentiate transcripts from each of two species participated within the syntrophic relationship, was recently employed to match the gene expression profiles of D. vulgaris in sulfate-limited monocultures and in syntrophic dual-cultures with a hydrogenotrophic methanogen Methanococcus maripaludis10 and of D. vulgaris during its metabolic shift from syntrophic growth with M. barkeri to sulfidogenic growth11. The results showed that between the 2 lifestyles, several hundred genes including those encoding ATPase, hydrogenases and high-molecular-weight cytochrome were differentially regulated, suggesting their potential roles to syntrophic growth relationship in D. vulgaris11,12. Interestingly, a gene cluster encoding several functionally unknown lipoproteins and membrane-bound proteins (DVU0145 to DVU0150) was found up-regulated in syntrophic dual-cultures when compared with the monocultures10 and down-regulated when D. vulgaris cells were shifted from syntrophic to sulfidogenic metabolism11, suggesting they may be involved in syntrophic metabolism. However, so far no further investigation on these genes have been conducted. Single-cell microbiology has attracted significant attention as more evidence suggested that even isogenic populations of microorganisms could have substantial cell-to-cell heterogeneity at both cellular and molecular levels12,13,14,15,16,17. For example, a RT-qPCR analysis of individual cells from the identical Escherichia coli population showed that the expression level of highly expressed the 16S rRNA gene could vary up to ~32-fold between single cells of the same population18. In addition to micro-scale environmental differences, it is currently known that gene-expression stochasticity, or noise, once amplified through generations, could eventually generate heterogeneity at the cellular level in a clonal bacterial population17,19,20. The significant gene-expression heterogeneity observed for a microbial population suggests that by simply harvesting and analyzing mRNA or proteins from whole populations, it may not be able to capture the unique patterns of gene expression related to distinct functional subpopulations. When it comes to mixed cultures, single-cell based analysis may be more valuable as the heterogeneity within a mixed population could be even higher as different types of cells with distinct metabolic profiles, interaction and stress responses, are co-cultivated within one culture21. Although single-cell genomics has been applied to a handful of symbiotic systems, including bacterial symbionts of marine sponges, insects (grasshoppers, termites)22, to our knowledge, the single-cell based gene-expression analysis has thus far not been applied to any syntrophic microbial system and therefore the dynamics of organic phenomenon and metabolic status in cells of syntrophic mixed cultures reminds unclear. In this work, we applied a single-cell RT-qPCR approach to reveal gene-expression heterogeneity during a model syntrophic system of Desulfovibrio vulgaris and Methanosarcina barkeri, as compared with the D. vulgaris monoculture. Using the optimized primers and single-cell analytical protocol, we quantitatively determine geneexpression levels of 6 selected target genes in each of the 120 single cells of D. vulgaris isolated from its monoculture and dualculture with M. barkeri. The results demonstrated very significant cell-to-cell gene-expression heterogeneity for the chosen D. vulgaris genes in both the monoculture and therefore the syntrophic dual-culture. Interestingly, no obvious increase in gene-expression heterogeneity for the chosen genes was observed for the syntrophic dual-culture in comparison with its monoculture, although the community structure and cell-cell interactions have become more complicated in the syntrophic dual-culture. In addition, the single-cell RT-qPCR analysis also provided further evidence that the gene cluster (DVU0148-DVU0150) could also be involved syntrophic metabolism between D. vulgaris and M. barkeri. Finally, the study validated that single-cell RT-qPCR analysis might be a valuable tool in deciphering gene functions and metabolism in mixed-cultured microbial communities.
PDFShare this article
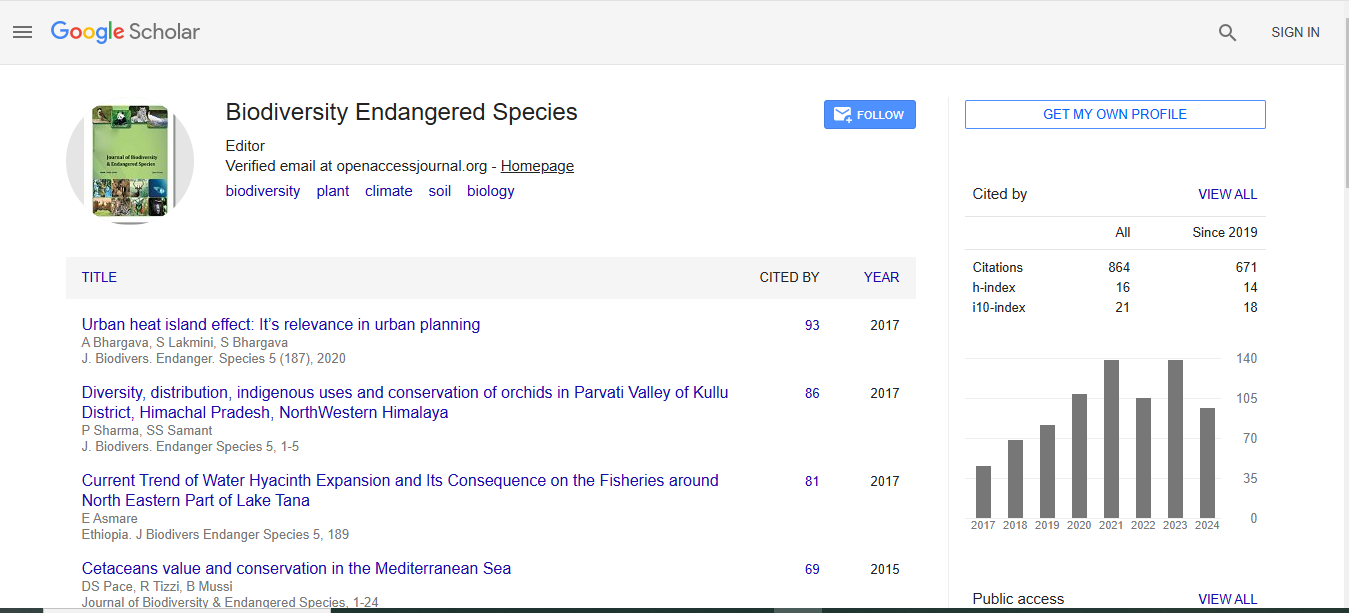
Journal of Biodiversity & Endangered Species received 624 citations as per Google Scholar report