GET THE APP
ISSN: 2155-9929
Sandhya Kille*
Share this article
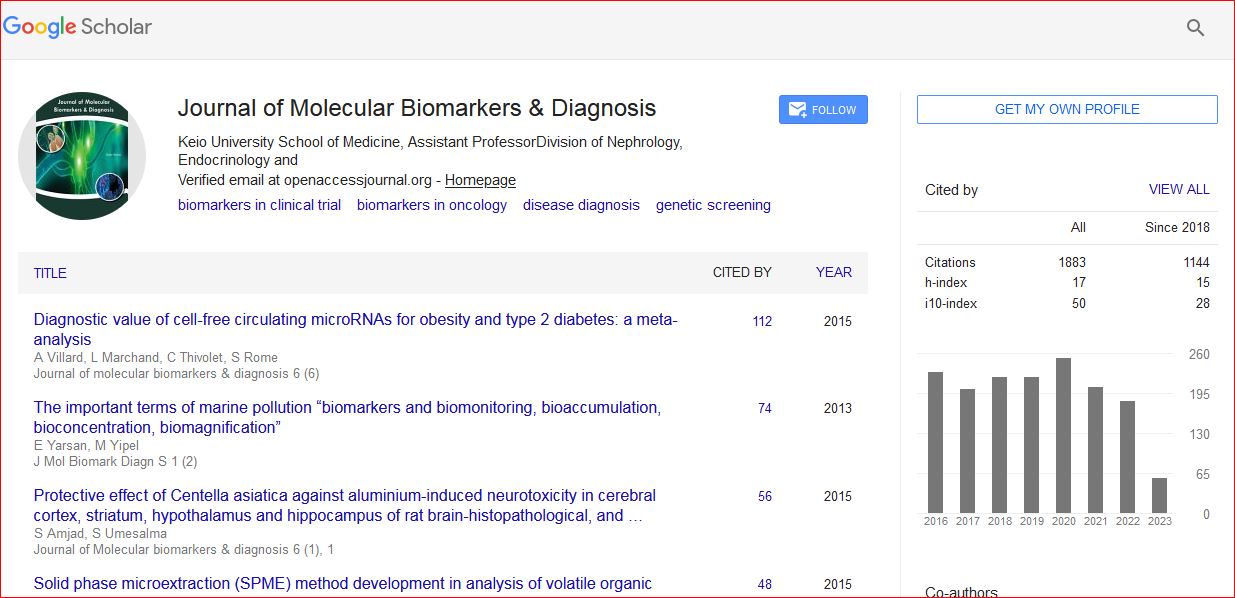
Journal of Molecular Biomarkers & Diagnosis received 2054 citations as per Google Scholar report