Perspective - (2021) Volume 8, Issue 9
Received: 04-Sep-2021
Published:
24-Sep-2021
, DOI: 10.37421/2476-2296.2021.8.201
Citation: Xiang Gao. "Areas of Application and Limits of Molecular Dynamics." Fluid Mech Open Acc 8 (2021): 201.
Copyright: © 2021 Xiang Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
MD is a pc simulation approach for analysing the physical actions of atoms and molecules. The atoms and molecules are allowed to engage for a hard and fast time period, giving a view of the dynamic "evolution" of the machine. Within the maximum not unusual version, the trajectories of atoms and molecules are decided with the aid of numerically solving Newton's equations of movement for a system of interacting debris, wherein forces among the debris and their potential energies are often calculated the use of interatomic potentials or molecular mechanics pressure fields. The method is implemented in the main in chemical physics, materials science, and biophysics. Because molecular structures commonly include a tremendous number of debris, it's miles impossible to decide the homes of such complicated structures analytically; MD simulation circumvents this problem by using the usage of numerical techniques. However, long MD simulations are mathematically unwell-conditioned, producing cumulative mistakes in numerical integration that can be minimized with right choice of algorithms and parameters, but no longer removed completely. For systems that obey the ergodic hypothesis, the evolution of 1 molecular dynamics simulation may be used to decide macroscopic thermodynamic properties of the machine: the time averages of an ergodic device correspond to micro canonical ensemble averages. MD has additionally been termed "statistical mechanics through numbers" and "Laplace's vision of Newtonian mechanics" of predicting the future by animating nature's forces and permitting insight into molecular motion on an atomic scale First used in theoretical physics, the MD technique gained popularity in substances technological know-how soon later on, and since the Seventies is likewise not unusual in biochemistry and biophysics. MD is frequently used to refine 3-dimensional structures of proteins and other macromolecules primarily based on experimental constraints from X-ray crystallography or NMR spectroscopy. In physics, MD is used to have a look at the dynamics of atomic-level phenomena that can't be observed without delay, which includes skinny-film boom and ion-sub plantation, and additionally to take a look at the bodily houses of Nano technological gadgets that have no longer or cannot but be created. In biophysics and structural biology, the technique is frequently applied to have a look at the motions of macromolecules consisting of proteins and nucleic acids, which may be useful for decoding the results of certain biophysical experiments and for modelling interactions with different molecules, as in ligand docking. The results of MD simulations may be examined thru contrast to experiments that measure molecular dynamics, of which a popular method is NMR spectroscopy. MD-derived shape predictions can be examined through network-wide experiments in critical assessment of protein structure Prediction (CASP), despite the fact that the technique has historically had limited success on this area. Michael Levitt, who shared the Nobel Prize in part for the utility of MD to proteins, wrote in 1999 that CASP participants typically did not use the method because of "... a principal embarrassment of molecular mechanics, particularly that electricity minimization or molecular dynamics normally ends in a version this is much less just like the experimental shape." improvements in computational resources allowing greater and longer MD trajectories, mixed with current enhancements in the first-class of pressure discipline parameters, have yielded some upgrades in each structure prediction and homology model refinement, without attaining the point of realistic utility in those areas; many pick out force area parameters as a key location for similarly development as an instance, Pinto et al. carried out MD simulations of Bcl-Xl complexes to calculate common positions of critical amino acids worried in ligand binding. however, Carlson et Snapshots of the protein at regular time durations during the simulation have been overlaid to perceive conserved binding regions (conserved in at least three out of eleven frames) for pharmacophore improvement. Spyrakis et al. depended on a workflow of MD simulations, finger prints for ligands and proteins (FLAP) and linear discriminate evaluation to pick out best ligand– protein conformations to behave as pharmacophore templates based totally on retrospective ROC evaluation of the ensuing pharmacophores. In an attempt to ameliorate structure-primarily based drug discovery modeling, the need for many modelled compounds, They proposed a mixture of MD simulation and ligand-receptor intermolecular contacts evaluation to parent important intermolecular contacts (binding interactions) from redundant ones in a unmarried ligand–protein complicated. vital contacts can then be transformed into pharmacophore fashions that can One run of an MD simulation optimizes the potential electricity, rather than the loose electricity of the protein that means that all entropic contributions to thermodynamic balance of protein structure are neglected, such as the conformational entropy of the polypeptide chain (the principle element that destabilizes protein shape) and hydrophobic effects (the main riding forces of protein folding). Some other vital thing is intramolecular hydrogen bonds, which aren't explicitly included in present day force fields, but described as Coulomb interactions of atomic factor prices. That is a crude approximation due to the fact hydrogen bonds have a partially quantum mechanical and chemical nature.
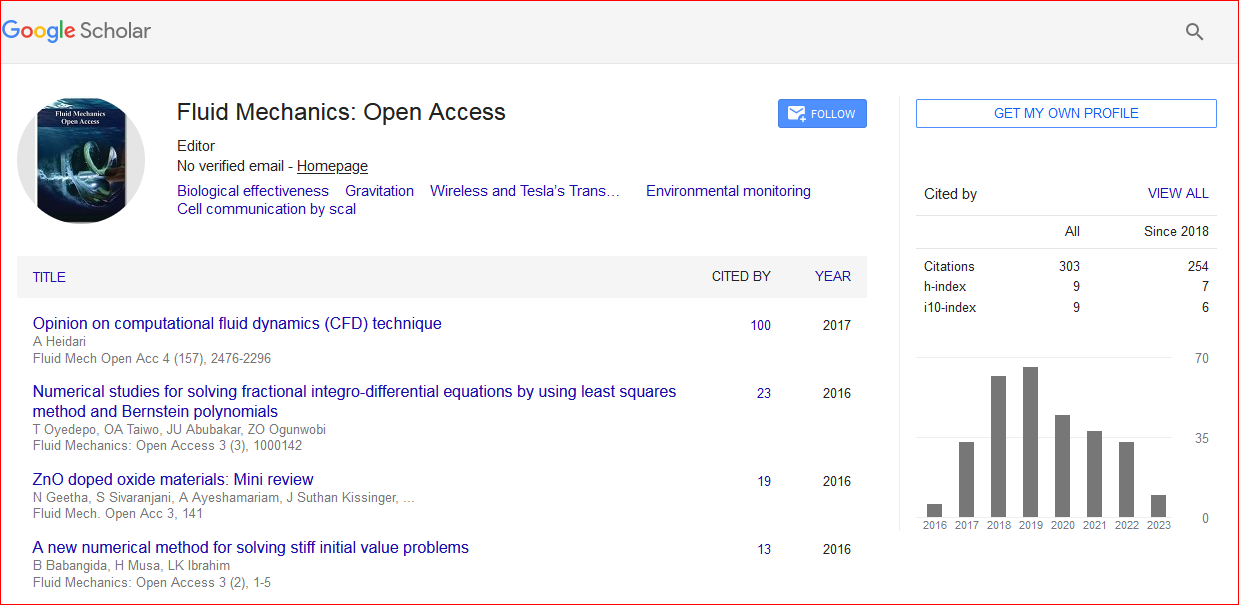
Fluid Mechanics: Open Access received 291 citations as per Google Scholar report